

Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders
- PMID: 20705090
- PMCID: PMC3167234
- DOI: 10.1016/j.pharmthera.2010.07.006
Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders
Abstract
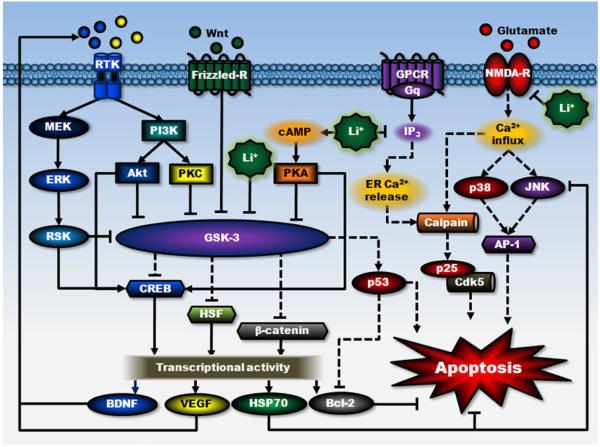
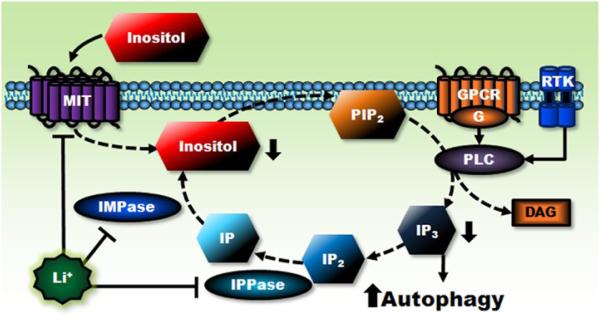
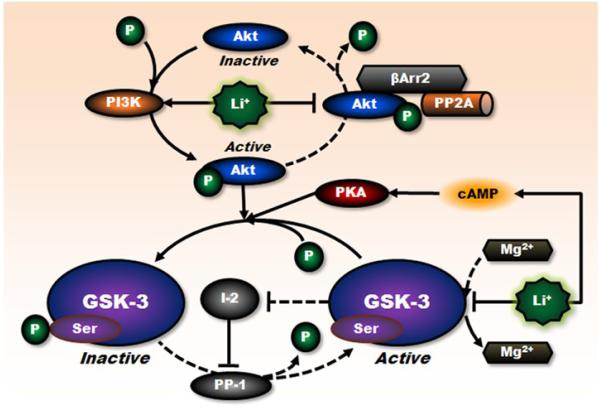
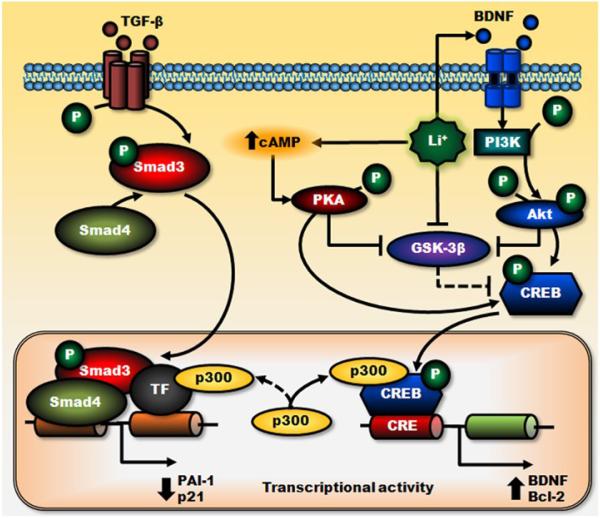
Lithium has been used clinically to treat bipolar disorder for over half a century, and remains a fundamental pharmacological therapy for patients with this illness. Although lithium's therapeutic mechanisms are not fully understood, substantial in vitro and in vivo evidence suggests that it has neuroprotective/neurotrophic properties against various insults, and considerable clinical potential for the treatment of several neurodegenerative conditions. Evidence from pharmacological and gene manipulation studies support the notion that glycogen synthase kinase-3 inhibition and induction of brain-derived neurotrophic factor-mediated signaling are lithium's main mechanisms of action, leading to enhanced cell survival pathways and alteration of a wide variety of downstream effectors. By inhibiting N-methyl-D-aspartate receptor-mediated calcium influx, lithium also contributes to calcium homeostasis and suppresses calcium-dependent activation of pro-apoptotic signaling pathways. In addition, lithium decreases inositol 1,4,5-trisphosphate by inhibiting phosphoinositol phosphatases, a process recently identified as a novel mechanism for inducing autophagy. Through these mechanisms, therapeutic doses of lithium have been demonstrated to defend neuronal cells against diverse forms of death insults and to improve behavioral as well as cognitive deficits in various animal models of neurodegenerative diseases, including stroke, amyotrophic lateral sclerosis, fragile X syndrome, as well as Huntington's, Alzheimer's, and Parkinson's diseases, among others. Several clinical trials are also underway to assess the therapeutic effects of lithium for treating these disorders. This article reviews the most recent findings regarding the potential targets involved in lithium's neuroprotective effects, and the implication of these findings for the treatment of a variety of diseases.
Published by Elsevier Inc.
Figures
Similar articles
-
Neuroprotective action of lithium in disorders of the central nervous system.Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011 Jun;36(6):461-76. doi: 10.3969/j.issn.1672-7347.2011.06.001. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011. PMID: 21743136 Free PMC article. Review.
-
Lithium neuroprotection: molecular mechanisms and clinical implications.Expert Rev Mol Med. 2004 Oct 18;6(21):1-18. doi: 10.1017/S1462399404008385. Expert Rev Mol Med. 2004. PMID: 15488156 Review.
-
Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: a role for neurotrophic and neuroprotective effects in manic depressive illness.J Clin Psychiatry. 2000;61 Suppl 9:82-96. J Clin Psychiatry. 2000. PMID: 10826666 Review.
-
Is There Justification to Treat Neurodegenerative Disorders by Repurposing Drugs? The Case of Alzheimer's Disease, Lithium, and Autophagy.Int J Mol Sci. 2020 Dec 27;22(1):189. doi: 10.3390/ijms22010189. Int J Mol Sci. 2020. PMID: 33375448 Free PMC article. Review.
-
Molecular mechanisms underlying mood stabilization in manic-depressive illness: the phenotype challenge.Am J Psychiatry. 1999 Oct;156(10):1506-14. doi: 10.1176/ajp.156.10.1506. Am J Psychiatry. 1999. PMID: 10518159 Review.
Cited by
-
Molecular mechanisms and therapeutic potential of lithium in Alzheimer's disease: repurposing an old class of drugs.Front Pharmacol. 2024 Jul 11;15:1408462. doi: 10.3389/fphar.2024.1408462. eCollection 2024. Front Pharmacol. 2024. PMID: 39055498 Free PMC article. Review.
-
An emerging role for the anti-inflammatory cytokine interleukin-10 in dengue virus infection.J Biomed Sci. 2013 Jun 25;20(1):40. doi: 10.1186/1423-0127-20-40. J Biomed Sci. 2013. PMID: 23800014 Free PMC article. Review.
-
What is the Role of Lithium in Epilepsy?Curr Neuropharmacol. 2022;20(10):1850-1864. doi: 10.2174/1570159X20666220411081728. Curr Neuropharmacol. 2022. PMID: 35410603 Free PMC article. Review.
-
The Concise Guide to PHARMACOLOGY 2013/14: enzymes.Br J Pharmacol. 2013 Dec;170(8):1797-867. doi: 10.1111/bph.12451. Br J Pharmacol. 2013. PMID: 24528243 Free PMC article.
-
Elucidating the pivotal molecular mechanisms, therapeutic and neuroprotective effects of lithium in traumatic brain injury.Brain Behav. 2024 Jun;14(6):e3595. doi: 10.1002/brb3.3595. Brain Behav. 2024. PMID: 38874089 Free PMC article. Review.
References
-
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. - PubMed
-
- Akassoglou K, Akpinar P, Murray S, Strickland S. Fibrin is a regulator of Schwann cell migration after sciatic nerve injury in mice. Neurosci.Lett. 2003;338:185–188. - PubMed
-
- Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr.Opin.Genet.Dev. 1998;8:55–62. - PubMed
-
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
