

Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NF{kappa}B cooperativity at adjacent response elements
- PMID: 20705611
- PMCID: PMC2951183
- DOI: 10.1074/jbc.M110.155309
Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NF{kappa}B cooperativity at adjacent response elements
Abstract
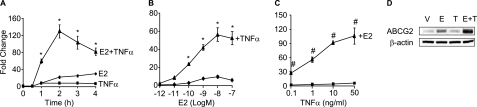
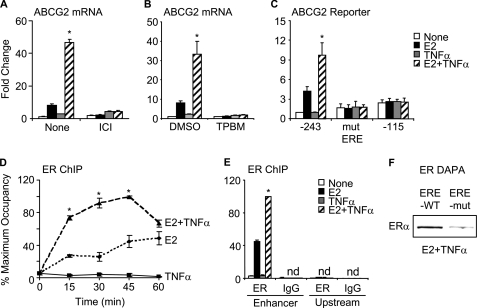
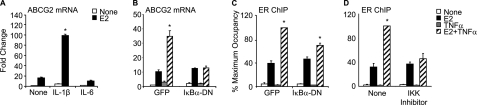
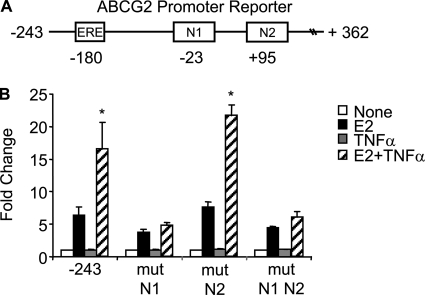
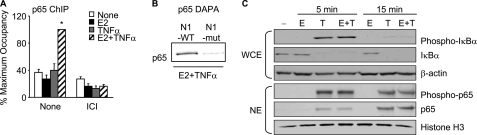
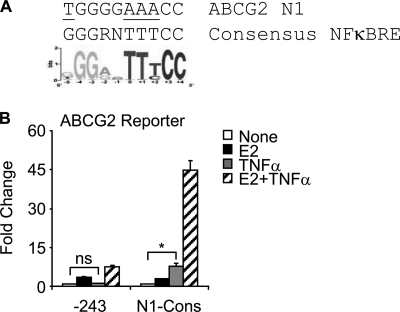
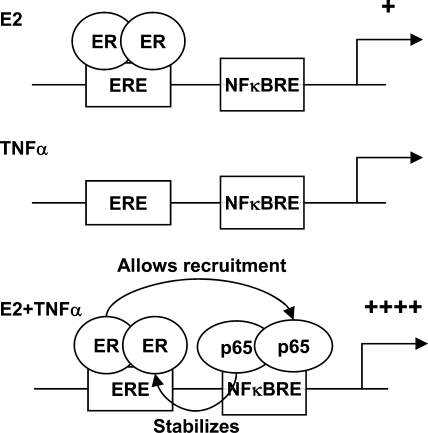
Constitutive activation of NFκB in estrogen receptor (ER)-positive breast cancer is associated with tumor recurrence and development of anti-estrogen resistance. Furthermore, a gene expression signature containing common targets for ER and NFκB has been identified and found to be associated with the more aggressive luminal B intrinsic subtype of ER-positive breast tumors. Here, we describe a novel mechanism by which ER and NFκB cooperate to up-regulate expression of one important gene from this signature, ABCG2, which encodes a transporter protein associated with the development of drug-resistant breast cancer. We and others have confirmed that this gene is regulated primarily by estrogen in an ER- and estrogen response element (ERE)-dependent manner. We found that whereas proinflammatory cytokines have little effect on this gene in the absence of 17β-estradiol, they can potentiate ER activity in an NFκB-dependent manner. ER allows the NFκB family member p65 to access a latent NFκB response element located near the ERE in the gene promoter. NFκB recruitment to the gene is, in turn, required to stabilize ER occupancy at the functional ERE. The result of this cooperative binding of ER and p65 at adjacent response elements leads to a major increase in both ABCG2 mRNA and protein expression. These findings indicate that estrogen and inflammatory factors can modify each other's activity through modulation of transcription factor accessibility and/or occupancy at adjacent response elements. This novel transcriptional mechanism could have important implications in breast cancer, where both inflammation and estrogen can promote cancer progression.
Figures
Similar articles
-
Transcriptional upregulation of breast cancer resistance protein by 17beta-estradiol in ERalpha-positive MCF-7 breast cancer cells.Oncology. 2006;71(5-6):446-55. doi: 10.1159/000108594. Epub 2007 Sep 17. Oncology. 2006. PMID: 17878748
-
Transcriptional modulation of BCRP gene to reverse multidrug resistance by toremifene in breast adenocarcinoma cells.Breast Cancer Res Treat. 2010 Oct;123(3):679-89. doi: 10.1007/s10549-009-0660-2. Epub 2009 Dec 6. Breast Cancer Res Treat. 2010. PMID: 19967559
-
[Regulation mechanism of breast cancer resistance protein by toremifene to reverse BCRP-mediated multidrug resistance in breast cancer cells].Zhonghua Zhong Liu Za Zhi. 2011 Sep;33(9):654-60. Zhonghua Zhong Liu Za Zhi. 2011. PMID: 22340044 Chinese.
-
NFκB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms.Mol Cell Endocrinol. 2015 Dec 15;418 Pt 3(0 3):235-9. doi: 10.1016/j.mce.2014.09.013. Epub 2014 Oct 18. Mol Cell Endocrinol. 2015. PMID: 25450861 Free PMC article. Review.
-
[Transcriptional regulation of breast cancer resistance protein].Yi Chuan. 2012 Dec;34(12):1529-36. doi: 10.3724/sp.j.1005.2012.01529. Yi Chuan. 2012. PMID: 23262099 Review. Chinese.
Cited by
-
Potentiating effects of GHRH analogs on the response to chemotherapy.Cell Cycle. 2015;14(5):699-704. doi: 10.1080/15384101.2015.1010893. Cell Cycle. 2015. PMID: 25648497 Free PMC article.
-
Apigenin by targeting hnRNPA2 sensitizes triple-negative breast cancer spheroids to doxorubicin-induced apoptosis and regulates expression of ABCC4 and ABCG2 drug efflux transporters.Biochem Pharmacol. 2020 Dec;182:114259. doi: 10.1016/j.bcp.2020.114259. Epub 2020 Oct 2. Biochem Pharmacol. 2020. PMID: 33011162 Free PMC article.
-
Antagonistic analogs of growth hormone-releasing hormone increase the efficacy of treatment of triple negative breast cancer in nude mice with doxorubicin; A preclinical study.Oncoscience. 2014 Oct 24;1(10):665-73. doi: 10.18632/oncoscience.92. eCollection 2014. Oncoscience. 2014. PMID: 25593995 Free PMC article.
-
Negative regulation of NF-κB by the ING4 tumor suppressor in breast cancer.PLoS One. 2012;7(10):e46823. doi: 10.1371/journal.pone.0046823. Epub 2012 Oct 4. PLoS One. 2012. PMID: 23056468 Free PMC article.
-
Role of ABCG2 expression driven by cisplatin in platinum-containing chemotherapy for gastric cancer.World J Gastroenterol. 2013 Oct 21;19(39):6630-6. doi: 10.3748/wjg.v19.i39.6630. World J Gastroenterol. 2013. PMID: 24151392 Free PMC article.
References
-
- Kushner P. J., Agard D. A., Greene G. L., Scanlan T. S., Shiau A. K., Uht R. M., Webb P. (2000) J. Steroid Biochem. Mol. Biol. 74, 311–317 - PubMed
-
- Kalaitzidis D., Gilmore T. D. (2005) Trends Endocrinol. Metab. 16, 46–52 - PubMed
-
- Bodine P. V., Harris H. A., Komm B. S. (1999) Endocrinology 140, 2439–2451 - PubMed
-
- Chu S., Nishi Y., Yanase T., Nawata H., Fuller P. J. (2004) Mol. Endocrinol. 18, 1919–1928 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
