

Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde
- PMID: 20705694
- PMCID: PMC3076664
- DOI: 10.1093/eurheartj/ehq253
Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde
Abstract
Aims: The present study was designed to examine the mechanism involved in mitochondrial aldehyde dehydrogenase (ALDH2)-induced cardioprotection against ischaemia/reperfusion (I/R) injury with a focus on autophagy.
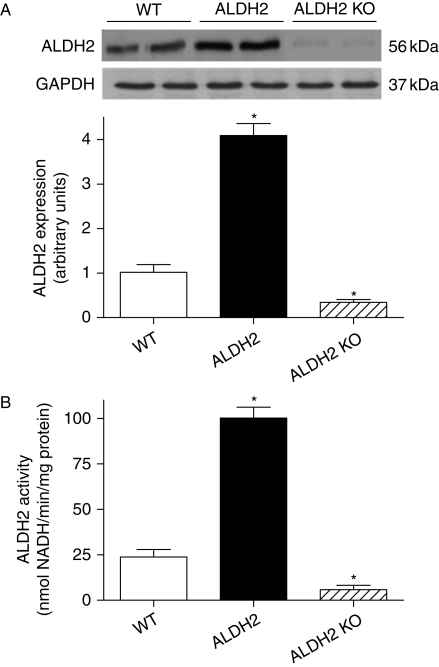
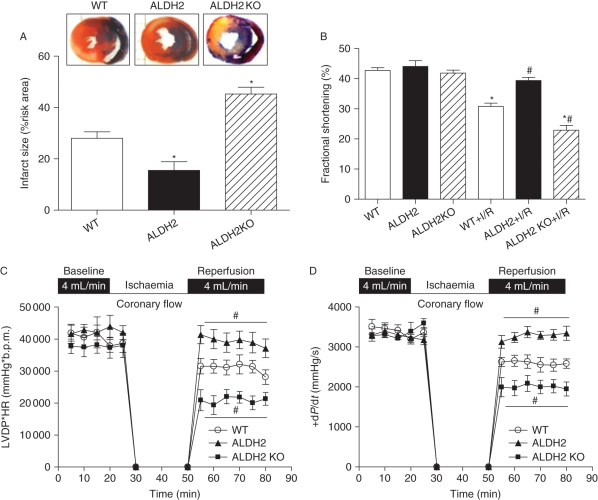
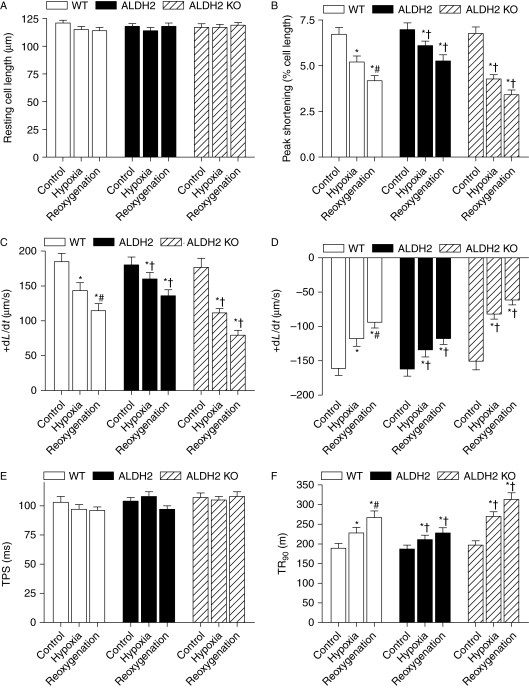
Methods: Wild-type (WT), ALDH2 overexpression, and knockout (KO) mice (n = 4-6 for each index measured) were subjected to I/R, and myocardial function was assessed using echocardiographic, Langendroff, and edge-detection systems. Western blotting was used to evaluate AMP-dependent protein kinase (AMPK), Akt, autophagy, and the AMPK/Akt upstream signalling LKB1 and PTEN.
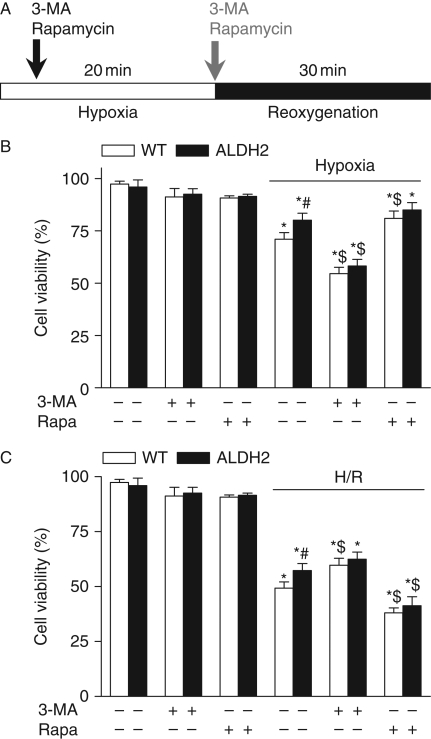
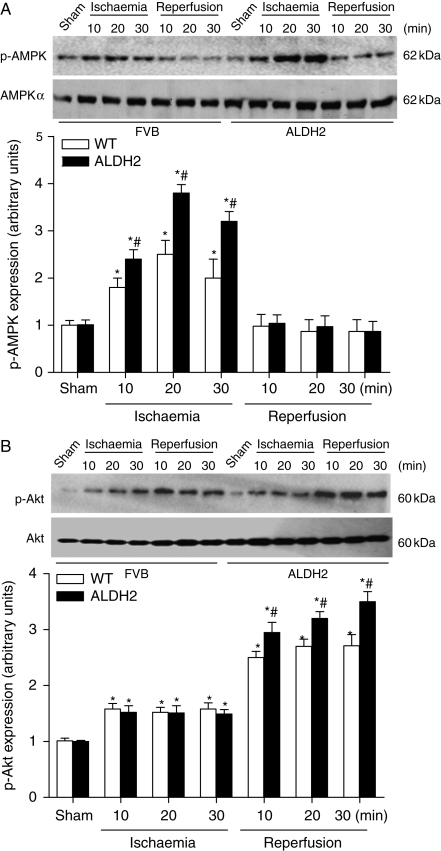
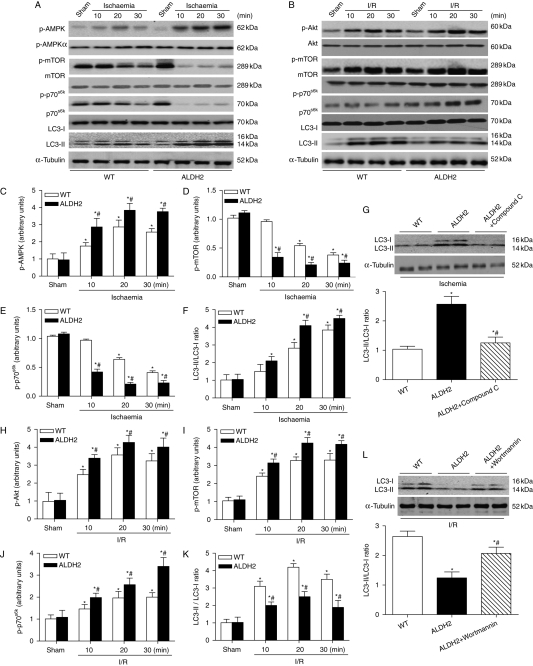
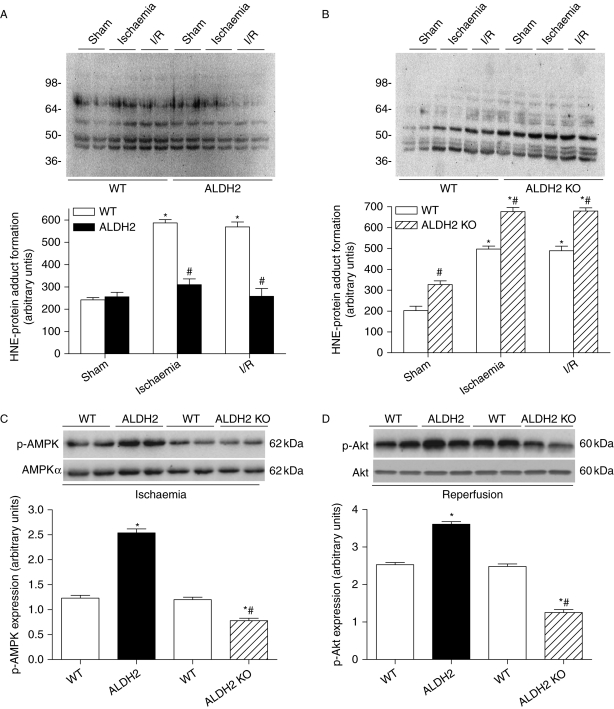
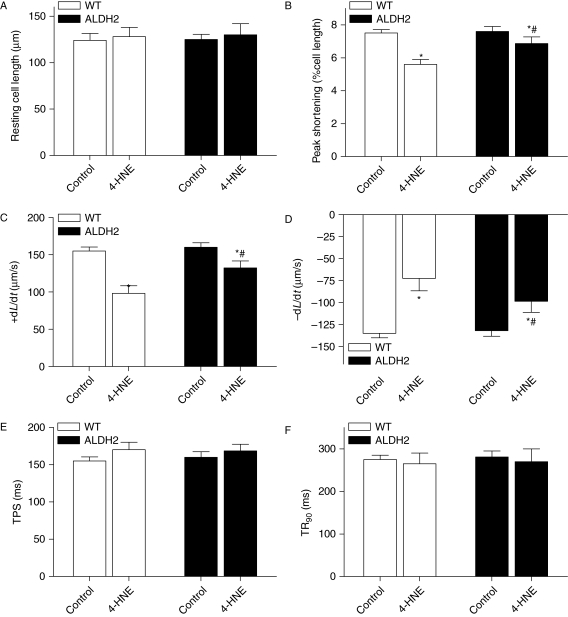
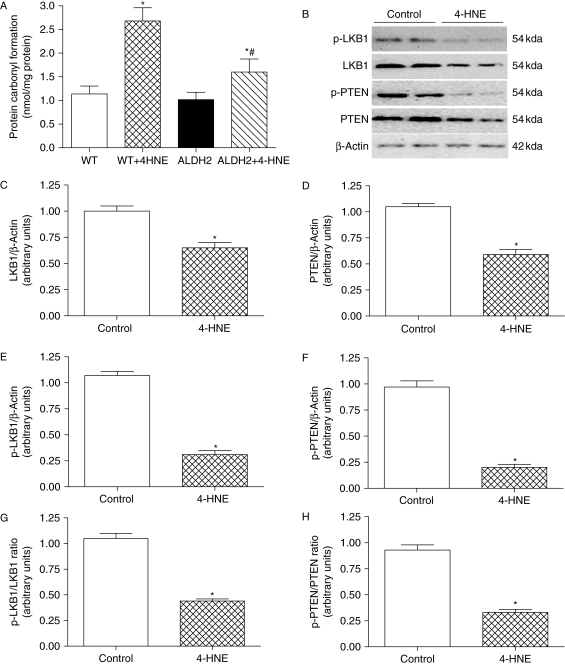
Results: ALDH2 overexpression and KO significantly attenuated and accentuated, respectively, infarct size, factional shortening, and recovery of post-ischaemic left ventricular function following I/R as well as hypoxia/reoxygenation-induced cardiomyocyte contractile dysfunction. Autophagy was induced during ischaemia and remained elevated during reperfusion. ALDH2 significantly promoted autophagy during ischaemia, which was accompanied by AMPK activation and mammalian target of rapamycin (mTOR) inhibition. On the contrary, ALDH2 overtly inhibited autophagy during reperfusion accompanied by the activation of Akt and mTOR. Inhibition and induction of autophagy mitigated ALDH2-induced protection against cell death in hypoxia and reoxygenation, respectively. In addition, levels of the endogenous toxic aldehyde 4-hydroxy-2-nonenal (4-HNE) were elevated by ischaemia and reperfusion, which was abrogated by ALDH2. Furthermore, ALDH2 ablated 4-HNE-induced cardiomyocyte dysfunction and protein damage, whereas 4-HNE directly decreased pan and phosphorylated LKB1 and PTEN expression.
Conclusion: Our data suggest a myocardial protective effect of ALDH2 against I/R injury possibly through detoxification of toxic aldehyde and a differential regulation of autophagy through AMPK- and Akt-mTOR signalling during ischaemia and reperfusion, respectively.
Figures
References
-
- Ren J. Acetaldehyde and alcoholic cardiomyopathy: lessons from the ADH and ALDH2 transgenic models. Novartis Found Symp. 2007;285:69–76. Discussion 76–69, 198–199. doi:10.1002/9780470511848.ch5. - DOI - PubMed
-
- Daiber A, Wenzel P, Oelze M, Munzel T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin Res Cardiol. 2008;97:12–20. doi:10.1007/s00392-007-0588-7. - DOI - PubMed
-
- Li SY, Li Q, Shen JJ, Dong F, Sigmon VK, Liu Y, Ren J. Attenuation of acetaldehyde-induced cell injury by overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene in human cardiac myocytes: role of MAP kinase signaling. J Mol Cell Cardiol. 2006;40:283–294. doi:10.1016/j.yjmcc.2005.11.006. - DOI - PubMed
-
- Li SY, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. J Mol Cell Cardiol. 2008;44:992–1001. doi:10.1016/j.yjmcc.2008.02.276. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
