

Review
doi: 10.1111/j.1600-0757.2010.00358.x.
Periodontal innate immune mechanisms relevant to atherosclerosis and obesity
- PMID: 20712641
- PMCID: PMC2928057
- DOI: 10.1111/j.1600-0757.2010.00358.x
Item in Clipboard
Review
Periodontal innate immune mechanisms relevant to atherosclerosis and obesity
Periodontol 2000.
2010 Oct.
No abstract available
Figures
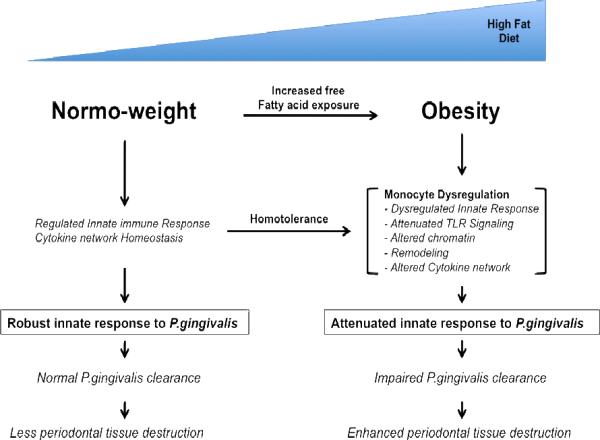
Model for proposed effect of obesity on innate response to P. gingivalis As exposure to free-fatty acids increases, common in obese individuals, this induces homotolerance along the Toll-like receptor-2 pathway in the innate immune system. Homotolerance alters Toll-like receptor signaling pathway by altering the expression levels of Toll-like receptor-2 and possibly chromatin remodeling at the Toll-like receptor-2 gene or other gene loci involved in the signaling pathway or cytokine release. The effect of the homotolerance leads to a dysregulated innate immune response and altered cytokine network upon exposure to P.gingivalis. As a result, the innate immune system has impaired clearance of P.gingivalis, which leads to enhancement of periodontal tissue destruction
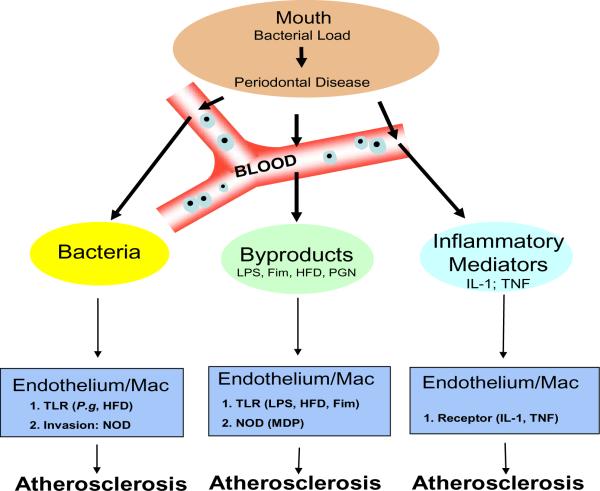
Relationship of P.gingivalis Infection and Atherosclerosis As the bacterial load of P.gingivalis in the oral cavity increases periodontal disease becomes more severe. However, damage is not just limited to the oral cavity since local vasculature enables an entry point for bacteria, bacterial by-products, or inflammatory mediators (e.g cytokines interleukin-1 or tumor necrosis factor-α). Bacteremias, common in periodontal disease due to dental work e.g. cleaning, flossing or surgery, effect both endothelium and macrophages by inducing homotolarance along the Toll-like receptor (TLR) pathway. However, since P.gingivalis is an invasive bacterium, mediated by its fimbriae (Fim) protein, it has the ability to activate the cytosolic nucleotide oligomerization domains (NODs) through recognition of muramyl dipeptide (MDP), a fragment of peptidoglycan (PGN). In addition, various bacterial by-products, e.g. Lipopolysaccharide (LPS), can also activate both the endothelium and macrophages. Further, from the oral cavity a state of local inflammation exists and immune cells are constantly releasing inflammatory mediators into the circulation, these mediators, interleukin-1 or tumor necrosis factor-α, can activate both the endothelium and macrophages. Finally, activated endothelium and macrophages have been shown to exacerbate Atherosclerosis when coupled with a high fat diet (HFD).
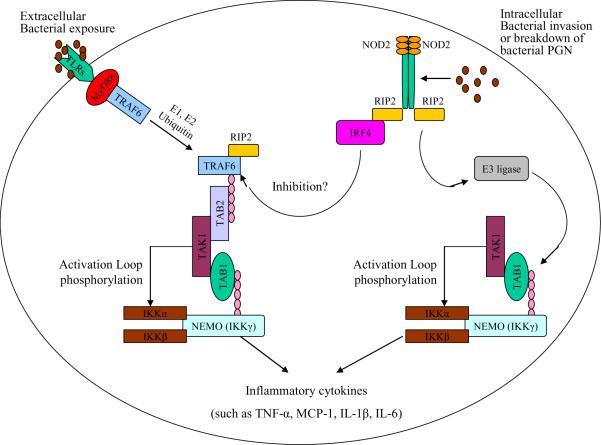
A system of pathogen recognition - Toll-Like Receptors (TLRs) and nucleotide oligomerization domain-2 (NOD2) signaling pathways Two primary bacterial pathogen sensors involved in P.ginigivalis recognition are the extracellular Toll-like Receptors (TLRs) and the intracellular nucleotide oligomerization domain-2 NOD2 receptors. TLRs bind to P.ginigvalis Lipopolysaccharide (LPS) and upon ligand binding recruit a number of adaptor molecules, e.g. tumor necrosis factor-α Receptor Associated Factor-6 (TRAF-6) and Myeloid differentiation primary response gene-88 (MyD88). This in turn ultimately triggers the polyubiquination, via E1 and E2 ubiqination, of NF-κB Essential Modulator (NEMO), also known as Inhibitor of NF-κB kinase-γ (IKK-γ), which in turn activates IKK-α and IKK-β subunits. IKK-α and IKK-β then phosphorylates Inhibitor of NF-κB (IκB) and promotes its degradation via the proteosome thus freeing NF-κB. The intracellular NOD2 receptor is activated upon binding to fragments of peptidoglycan (PGN), muramyl dipeptide (MDP), which then recruits Receptor Interacting Protein-2 (RIP2), a serine-theronine kinase. The NOD2/RIP2 complex undergoes oligomerization itself, which enables direct binding to NEMO (IKK-γ). NOD2/RIP2/NEMO complex can then recruit E3-Ligase and promote the polyubiquination of IKK-α and, via a similar mechanism as TLR signaling, activates NF-κB. Ultimately, both TLRs and NOD2 signaling converge and result in NF-κB translocating into the nucleus and increase transcription of pro-inflammatory cytokines (e.g. tumor necrosis factor-α, MCP-1, interleukin-1β, and interleukin-6).
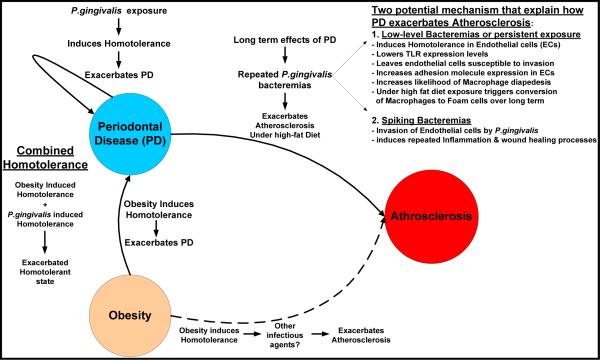
Proposed integrated model linking Periodontal Disease, Obesity, and Atherosclerosis We propose the following integrated model linking Periodontal Disease, Obesity and Atherosclerosis with a foundation built on a foundation of dysregulated innate immunity due to the induction of homotolerance. Homotolerance can be induced either by obesity directly or by exposure to P.gingivalis. However, when obesity is combined with P.gingivalis exposure, we speculated that exacerbates the overall homotolerant state thus having an additive effect and further exacerbating Periodontal Disease. We speculate two potential mechanisms that explain how the long-term consequences of periodontal disease exacerbates Atherosclerosis. First is the induction of homotolerance through a persistent low-level bacteremia or perhaps a transient low-level bacteremia induces a tolerant state within the endothelial cells and Macrophages centered on regions of fatty-streaks/athroma development. Specifically, this results in reduced expression of Toll-like Receptors (TLR), which then leaves these cells susceptible to P. gingivalis invasion and increased adhesion receptor expression via nucleotide oligomerization domain-2 NOD2 receptor signaling. Increased adhesion receptor expression increases the likelihood of macrophages transiting from the blood compartment to the aterial intima. When coupled with a high-fat diet macrophages will uptake cholesterol from Low-desnity lipoprotein via the scavenger receptor. Over time this has been shown to foster conversion of the macrophage to a `Foam' cell and progression of Atherosclerosis. Secondly, there is a potential for a spiking bactermia during dental procedures or flossing that could actually overcome the homotolerance and result in a more traditional inflammatory response and wound healing processes which have been shown to exacerbate Atherosclerosis.
References
-
- Aderem A. Phagocytosis and the inflammatory response. J infect dis. 2003;187(Suppl 2):S340–345. - PubMed
-
- Akira S, Uematsu S S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. - PubMed
-
- Amabile N, Susini G, Pettenati-Soubayroux I, Bonello L, Gil JM, Arques S, Bonfil JJ, Paganelli F. Severity of periodontal disease correlates to inflammatory systemic status and independently predicts the presence and angiographic extent of stable coronary artery disease. J Intern Med. 2008;263:644–652. - PubMed
-
- Amar S, Han X. The impact of periodontal infection on systemic diseases. Med Sci Monit. 2003;9:RA291–299. - PubMed
-
- Amar S, Wu SC, Madan M. Is Porphyromonas gingivalis cell invasion required for atherogenesis? Pharmacotherapeutic implications. J Immunol. 2009;182:1584–1592. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
