

Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease
- PMID: 20713516
- PMCID: PMC2922500
- DOI: 10.1101/gad.1958410
Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease
Abstract
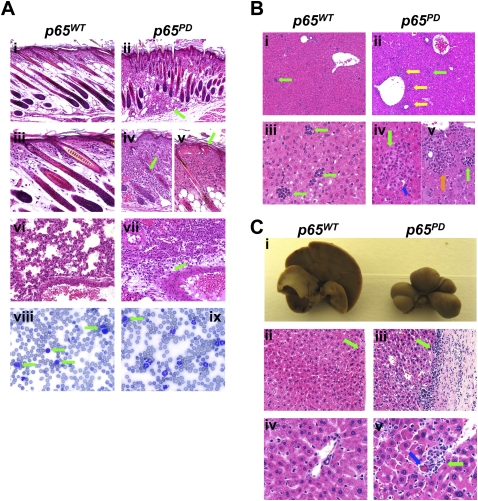
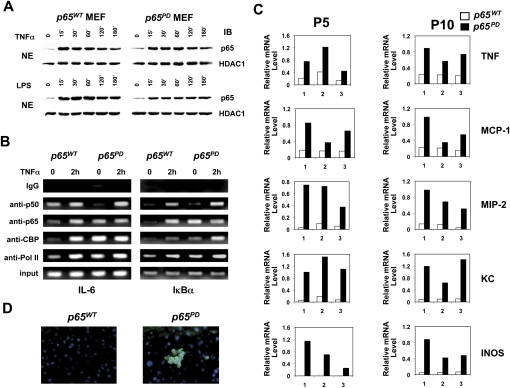
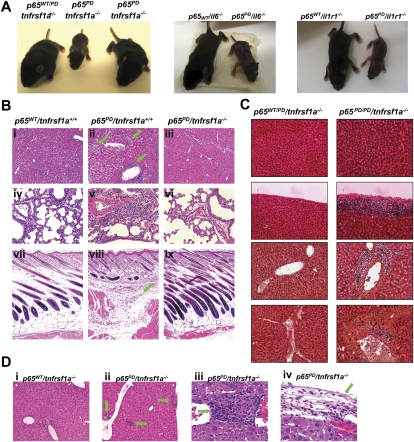
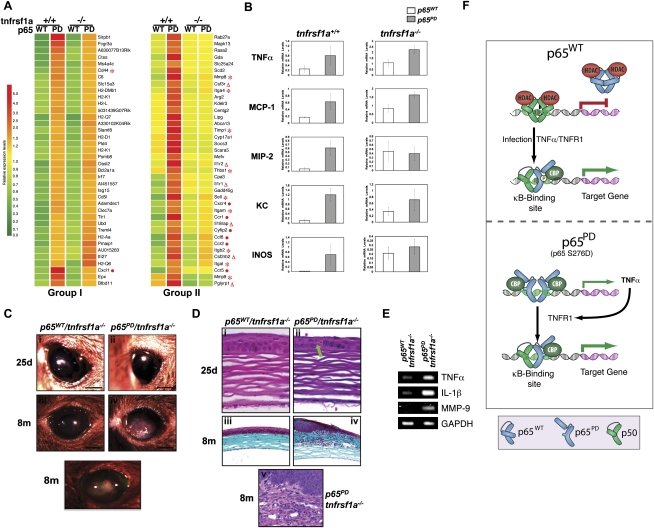
NF-kappaB is well established as a key component of the inflammatory response. However, the precise mechanisms through which NF-kappaB activation contributes to inflammatory disease states remain poorly defined. To test the role of NF-kappaB in inflammation, we created a knock-in mouse that expresses a constitutively active form of NF-kappaB p65 dimers. These mice are born at normal Mendelian ratios, but display a progressive, systemic hyperinflammatory condition that results in severe runting and, typically, death 8-20 d after birth. Examination of homozygous knock-in mice demonstrates significant increases in proinflammatory cytokines and chemokines. Remarkably, crossing this strain with mice lacking TNF receptor 1 (TNFR1) leads to a complete rescue of the hyperinflammatory phenotype. However, upon aging, these rescued mice begin to display chronic keratitis accompanied by increased corneal expression of TNFalpha, IL-1beta, and MMP-9, similar to that seen in human keratoconjunctivitis sicca (KCS) or "dry eyes." Therefore, our results show that, while constitutively active NF-kappaB can trigger systemic inflammation, it does so indirectly, through increased TNF production. However, certain inflammatory disease states, such as keratitis or KCS, a condition that is seen in Sjogren's syndrome, are dependent on NF-kappaB, but are independent of TNFR1 signaling.
Figures

References
-
- Anest V, Cogswell PC, Baldwin AS Jr 2004. IκB kinase α and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IκBα degradation. J Biol Chem 279: 31183–31189 - PubMed
-
- Chen LF, Greene WC 2004. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol 5: 392–401 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous