

Foxp1 coordinates cardiomyocyte proliferation through both cell-autonomous and nonautonomous mechanisms
- PMID: 20713518
- PMCID: PMC2922503
- DOI: 10.1101/gad.1929210
Foxp1 coordinates cardiomyocyte proliferation through both cell-autonomous and nonautonomous mechanisms
Abstract
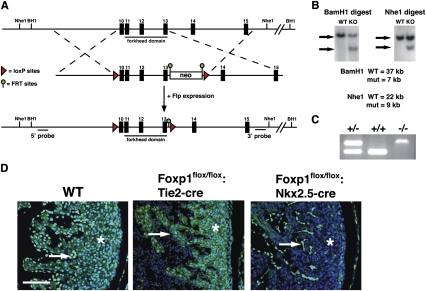
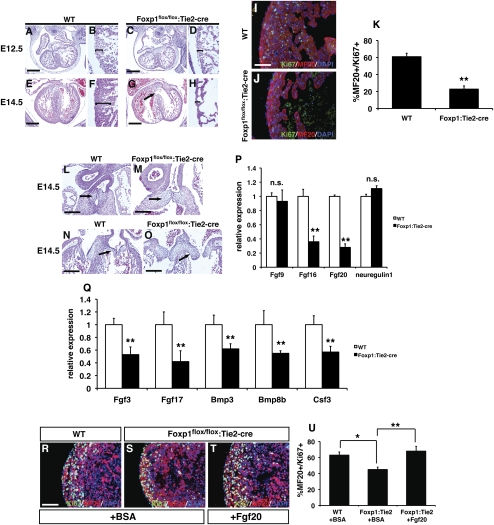
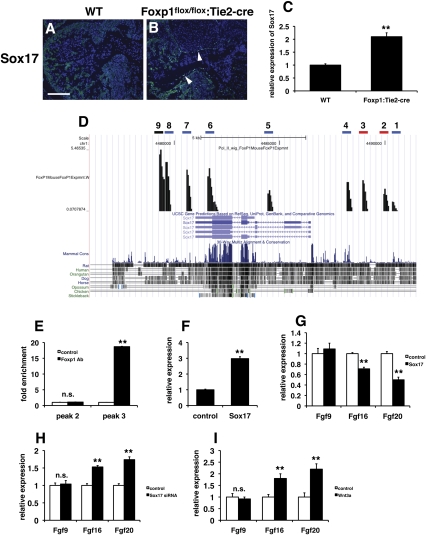
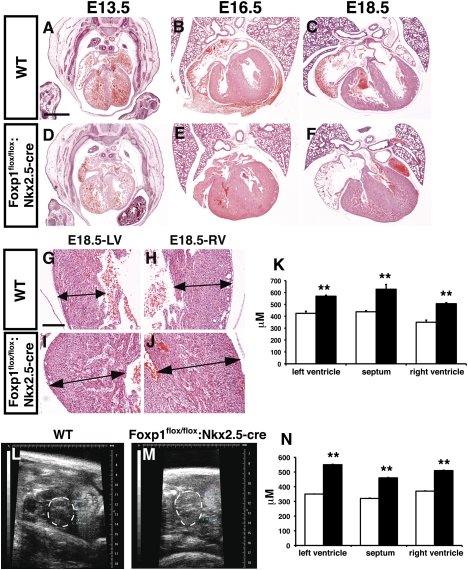
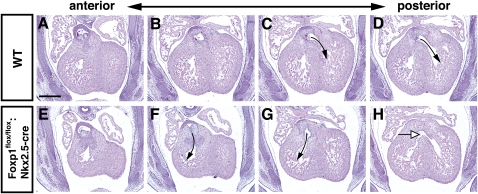
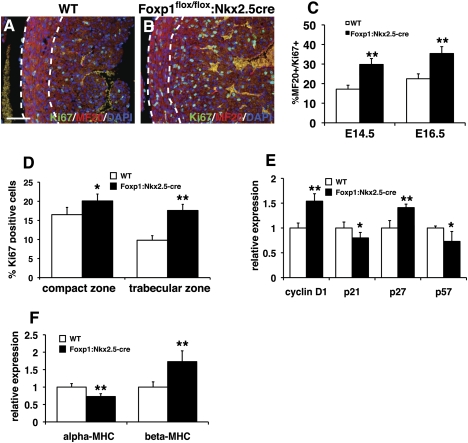
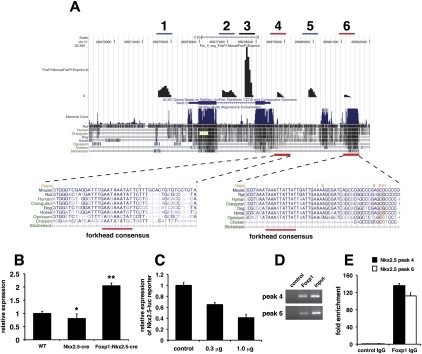
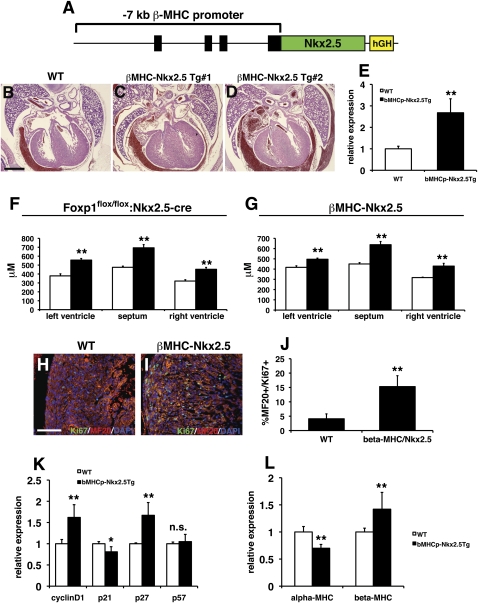
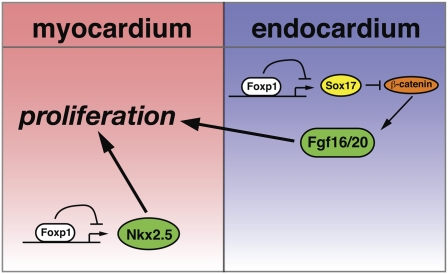
Cardiomyocyte proliferation is high in early development and decreases progressively with gestation, resulting in the lack of a robust cardiomyocyte proliferative response in the adult heart after injury. Little is understood about how both cell-autonomous and nonautonomous signals are integrated to regulate the balance of cardiomyocyte proliferation during development. In this study, we show that a single transcription factor, Foxp1, can control the balance of cardiomyocyte proliferation during development by targeting different pathways in the endocardium and myocardium. Endocardial loss of Foxp1 results in decreased Fgf3/Fgf16/Fgf17/Fgf20 expression in the heart, leading to reduced cardiomyocyte proliferation. This loss of myocardial proliferation can be rescued by exogenous Fgf20, and is mediated, in part, by Foxp1 repression of Sox17. In contrast, myocardial-specific loss of Foxp1 results in increased cardiomyocyte proliferation and decreased differentiation, leading to increased myocardial mass and neonatal demise. We show that Nkx2.5 is a direct target of Foxp1 repression, and Nkx2.5 expression is increased in Foxp1-deficient myocardium. Moreover, transgenic overexpression of Nkx2.5 leads to increased cardiomyocyte proliferation and increased ventricular mass, similar to the myocardial-specific loss of Foxp1. These data show that Foxp1 coordinates the balance of cardiomyocyte proliferation and differentiation through cell lineage-specific regulation of Fgf ligand and Nkx2.5 expression.
Figures
Similar articles
-
Regulation of cardiomyocyte proliferation by Foxp1.Cell Cycle. 2010 Nov 1;9(21):4251-2. doi: 10.4161/cc.9.21.13924. Epub 2010 Nov 12. Cell Cycle. 2010. PMID: 21051948 No abstract available.
-
Genetic abnormalities in FOXP1 are associated with congenital heart defects.Hum Mutat. 2013 Sep;34(9):1226-30. doi: 10.1002/humu.22366. Epub 2013 Jul 11. Hum Mutat. 2013. PMID: 23766104 Free PMC article.
-
Endothelial Forkhead Box Transcription Factor P1 Regulates Pathological Cardiac Remodeling Through Transforming Growth Factor-β1-Endothelin-1 Signal Pathway.Circulation. 2019 Aug 20;140(8):665-680. doi: 10.1161/CIRCULATIONAHA.119.039767. Epub 2019 Jun 10. Circulation. 2019. PMID: 31177814
-
FOXP1: a potential therapeutic target in cancer.Expert Opin Ther Targets. 2007 Jul;11(7):955-65. doi: 10.1517/14728222.11.7.955. Expert Opin Ther Targets. 2007. PMID: 17614763 Free PMC article. Review.
-
Targeting the forkhead box protein P1 pathway as a novel therapeutic approach for cardiovascular diseases.Heart Fail Rev. 2022 Jan;27(1):345-355. doi: 10.1007/s10741-020-09992-2. Heart Fail Rev. 2022. PMID: 32648149 Review.
Cited by
-
FOXP1 potentiates Wnt/β-catenin signaling in diffuse large B cell lymphoma.Sci Signal. 2015 Feb 3;8(362):ra12. doi: 10.1126/scisignal.2005654. Sci Signal. 2015. PMID: 25650440 Free PMC article.
-
FoxO1 and FoxM1 transcription factors have antagonistic functions in neonatal cardiomyocyte cell-cycle withdrawal and IGF1 gene regulation.Circ Res. 2013 Jan 18;112(2):267-77. doi: 10.1161/CIRCRESAHA.112.277442. Epub 2012 Nov 14. Circ Res. 2013. PMID: 23152492 Free PMC article.
-
Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function.Nat Immunol. 2019 Feb;20(2):232-242. doi: 10.1038/s41590-018-0291-z. Epub 2019 Jan 14. Nat Immunol. 2019. PMID: 30643266 Free PMC article.
-
FOXP1, an estrogen-inducible transcription factor, modulates cell proliferation in breast cancer cells and 5-year recurrence-free survival of patients with tamoxifen-treated breast cancer.Horm Cancer. 2011 Oct;2(5):286-97. doi: 10.1007/s12672-011-0082-6. Horm Cancer. 2011. PMID: 21901488 Free PMC article.
-
Opposing roles of FoxP1 and Nfat3 in transcriptional control of cardiomyocyte hypertrophy.Mol Cell Biol. 2011 Jul;31(14):3068-80. doi: 10.1128/MCB.00925-10. Epub 2011 May 23. Mol Cell Biol. 2011. Retraction in: Mol Cell Biol. 2020 Dec 21;41(1):e00544-20. doi: 10.1128/MCB.00544-20. PMID: 21606195 Free PMC article. Retracted.
References
-
- Bersell K, Arab S, Haring B, Kuhn B 2009. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138: 257–270 - PubMed
-
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27: 68–73 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases