

Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis
- PMID: 20716917
- PMCID: PMC2835918
- DOI: 10.4161/oxim.2.5.9541
Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis
Abstract
Background: Oxidative stress and inflammation may contribute to the disruption of the protective gut barrier through various mechanisms; mitochondrial dysfunction resulting from inflammatory and oxidative injury may potentially be a significant source of apoptosis during necrotizing enterocolitis (NEC). Tumor necrosis factor (TNF)-alpha is thought to generate reactive oxygen species (ROS) and activate the apoptosis signal-regulating kinase 1 (ASK1)-c-Jun N-terminal kinase (JNK)/p38 pathway. Hence, the focus of our study was to examine the effects of TNF-alpha/ROS on mitochondrial function, ASK1-JNK/p38 cascade activation in intestinal epithelial cells during NEC.
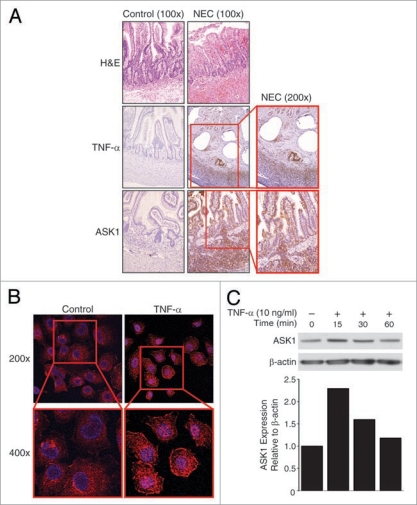
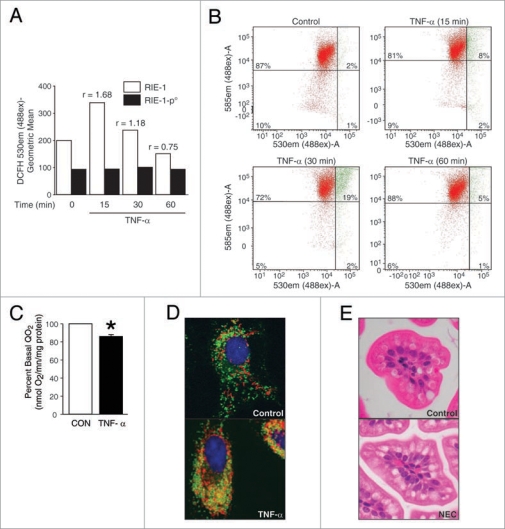
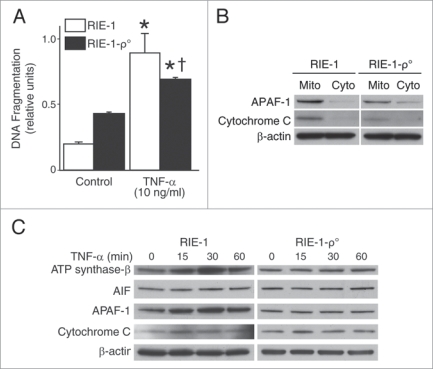
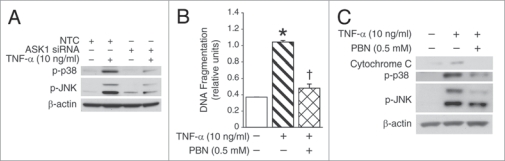
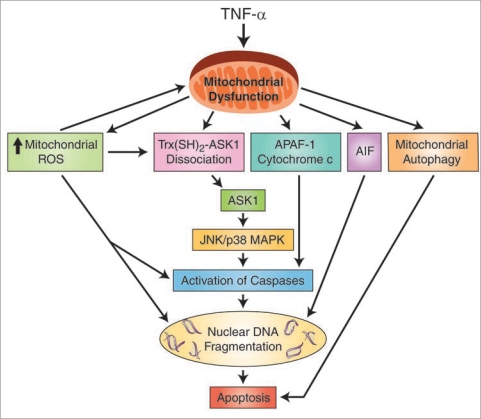
Results: We found (a) abundant tissue TNF-alpha and ASK1 expression throughout all layers of the intestine in neonates with NEC, suggesting that TNF-alpha/ASK1 may be a potential source (indicators) of intestinal injury in neonates with NEC; (b) TNF-alpha-induced rapid and transient activation of JNK/p38 apoptotic signaling in all cell lines suggests that this may be an important molecular characteristic of NEC; (c) TNF-alpha-induced rapid and transient ROS production in RIE-1 cells indicates that mitochondria are the predominant source of ROS, demonstrated by significantly attenuated response in mitochondrial DNA-depleted (RIE-1-rho) intestinal epithelial cells; (d) further studies with mitochondria-targeted antioxidant PBN supported our hypothesis that effective mitochondrial ROS trapping is protective against TNF-alpha/ROS-induced intestinal epithelial cell injury; (e) TNF-alpha induces significant mitochondrial dysfunction in intestinal epithelial cells, resulting in increased production of mtROS, drop in mitochondrial membrane potential (MMP) and decreased oxygen consumption; (f) although the significance of mitochondrial autophagy in NEC has not been unequivocally shown, our studies provide a strong preliminary indication that TNF-alpha/ROS-induced mitochondrial autophagy may play a role in NEC, and this process is a late phenomenon.
Methods: Paraffin-embedded intestinal sections from neonates with NEC and non-inflammatory condition of the gastrointestinal tract undergoing bowel resections were analyzed for TNF-alpha and ASK1 expression. Rat (RIE-1) and mitochondrial DNA-depleted (RIE-1-rho) intestinal epithelial cells were used to determine the effects of TNF-alpha on mitochondrial function.
Conclusions: Our findings suggest that TNF-alpha induces significant mitochondrial dysfunction and activation of mitochondrial apoptotic responses, leading to intestinal epithelial cell apoptosis during NEC. Therapies directed against mitochondria/ROS may provide important therapeutic options, as well as ameliorate intestinal epithelial cell apoptosis during NEC.
Figures
References
-
- Kelly N, Friend K, Boyle P, Zhang XR, Wong C, Hackam DJ, et al. The role of the glutathione antioxidant system in gut barrier failure in a rodent model of experimental necrotizing enterocolitis. Surgery. 2004;136:557–566. - PubMed
-
- Martin CR, Walker WA. Intestinal immune defences and the inflammatory response in necrotising enterocolitis. Semin Fetal Neonatal Med. 2006;11:369–377. - PubMed
-
- Markel TA, Crisostomo PR, Wairiuko GM, Pitcher J, Tsai BM, Meldrum DR. Cytokines in necrotizing enterocolitis. Shock. 2006;25:329–337. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
