

Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches
- PMID: 20716935
- PMCID: PMC2952095
- DOI: 10.4161/oxim.3.2.11354
Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches
Abstract
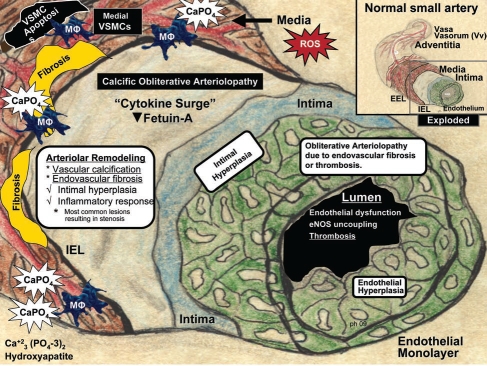
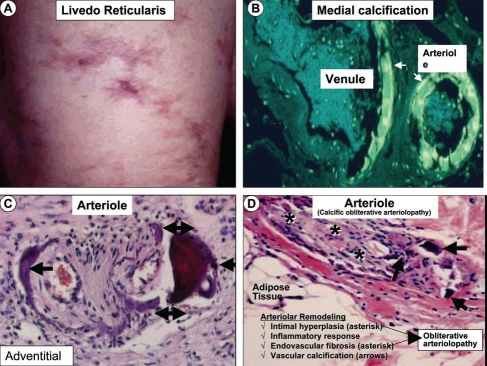
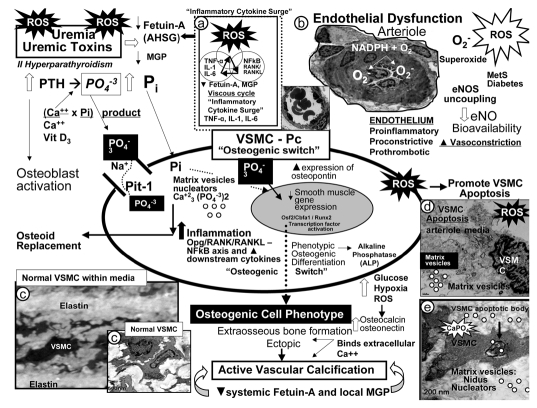
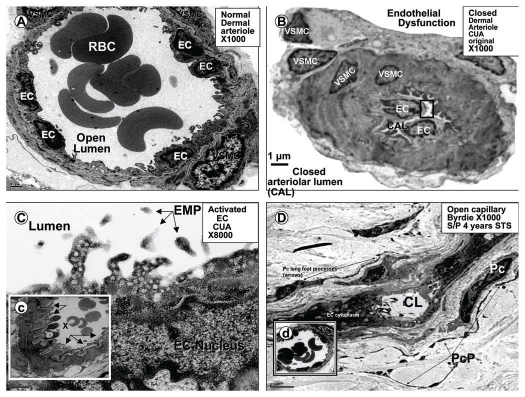
Calcific uremic arteriolopathy (CUA)/calciphylaxis is an important cause of morbidity and mortality in patients with chronic kidney disease requiring renal replacement. Once thought to be rare, it is being increasingly recognized and reported on a global scale. The uremic milieu predisposes to multiple metabolic toxicities including increased levels of reactive oxygen species and inflammation. Increased oxidative stress and inflammation promote this arteriolopathy by adversely affecting endothelial function resulting in a prothrombotic milieu and significant remodeling effects on vascular smooth muscle cells. These arteriolar pathological effects include intimal hyperplasia, inflammation, endovascular fibrosis and vascular smooth muscle cell apoptosis and differentiation into bone forming osteoblast-like cells resulting in medial calcification. Systemic factors promoting this vascular condition include elevated calcium, parathyroid hormone, and hyperphosphatemia with consequent increases in the calcium x phosphate product. The uremic milieu contributes to a marked increased in upstream reactive oxygen species - oxidative stress and subsequent downstream increased inflammation, in part, via activation of the nuclear transcription factor NFkappaB and associated downstream cytokine pathways. Consitutive anti-calcification proteins such as Fetuin-A and matrix GLA proteins and their signaling pathways may be decreased, which further contributes to medial vascular calcification. The resulting clinical entity is painful, debilitating and contributes to the excess morbidity and mortality associated with chronic kidney disease and end stage renal disease. These same histopathologic conditions also occur in patients without uremia and therefore, the term calcific obliterative arteriolopathy could be utilized in these conditions.
Figures




References
-
- Coates T, Kirkland GS, Dymock RB, Murphy BF, Brealey JK, Mathew TH, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384–391. - PubMed
-
- Pollock B, Cunliffe W, Merchant W. Calciphylaxis in the absence of renal failure. Clin Exp Dermatol. 2000;25:389. - PubMed
-
- Goyal S, Huhn K, Provost T. Calciphylaxis in a patient without renal failure or elevated parathyroid hormone: the possible aetiological role of chemotherapy. Br J Dermatol. 2000;143:1087–1090. - PubMed
-
- Don BR, Chin AL. A strategy for the treatment of calcific uremic arteriolopathy (calciphylaxis) employing a combination of therapies. Clin Nephrol. 2003;59:463–470. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
