

Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways
- PMID: 20716939
- PMCID: PMC2952099
- DOI: 10.4161/oxim.3.2.11758
Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways
Abstract
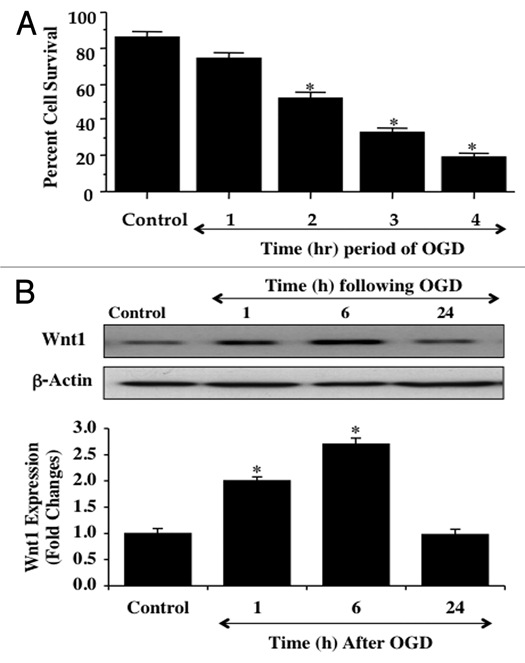
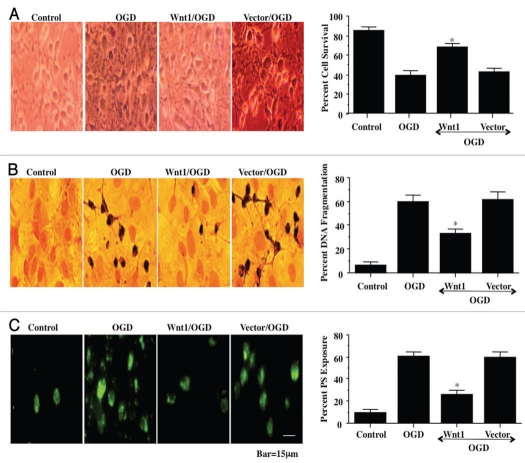
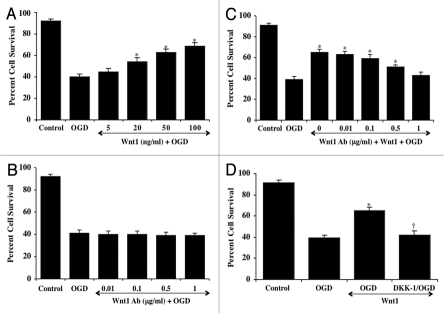
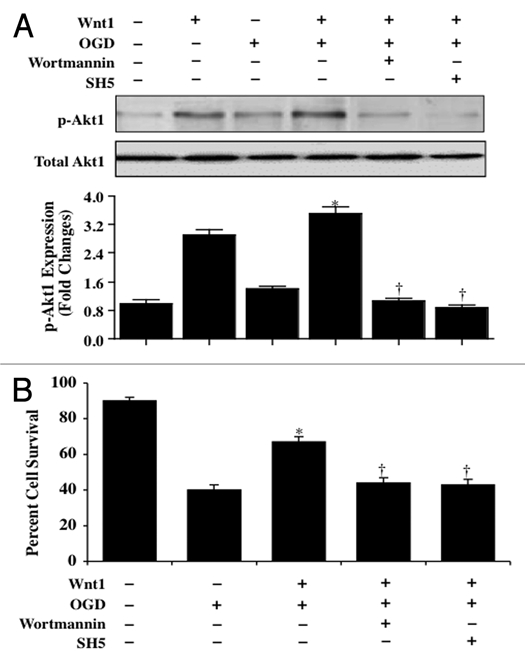
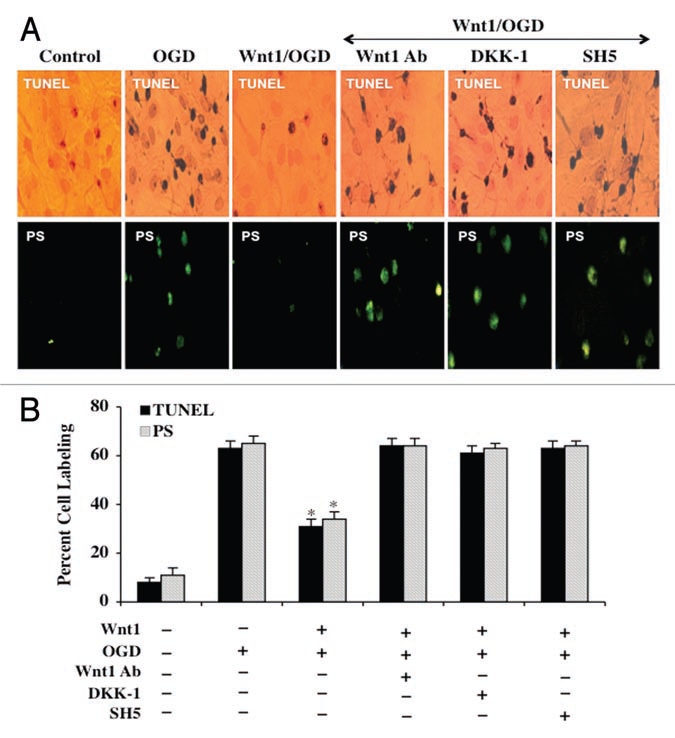
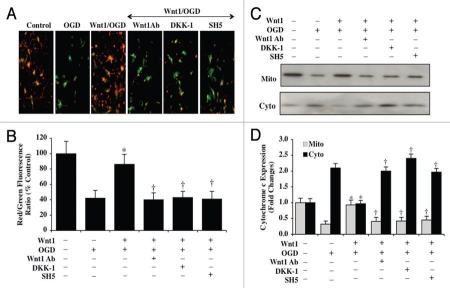
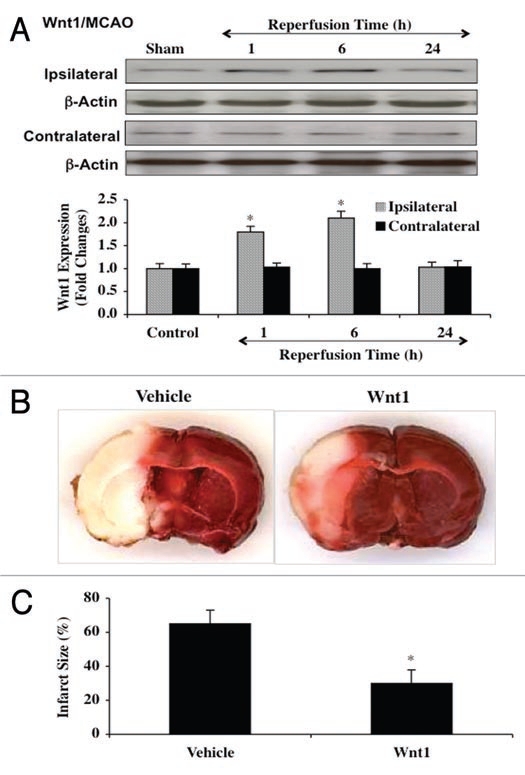
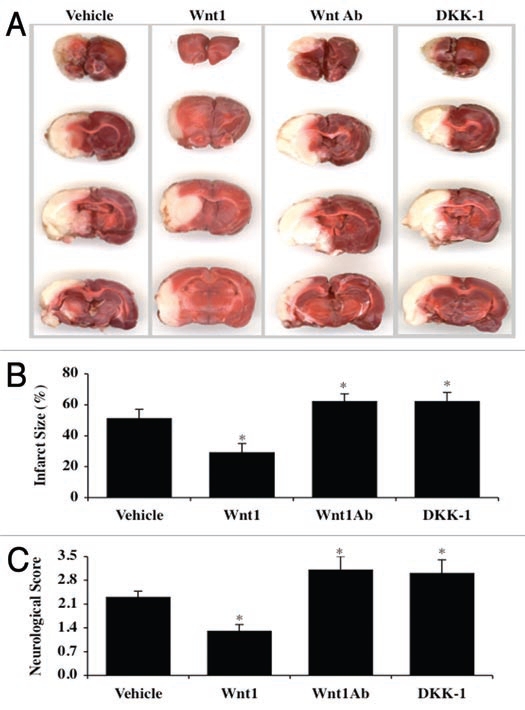
Although essential for the development of the nervous system, Wnt1 also has been associated with neurodegenerative disease and cognitive loss during periods of oxidative stress. Here we show that endogenous expression of Wnt1 is suppressed during oxidative stress in both in vitro and in vivo experimental models. Loss of endogenous Wnt1 signaling directly correlates with neuronal demise and increased functional deficit, illustrating that endogenous neuronal Wnt1 offers a vital level of intrinsic cellular protection against oxidative stress. Furthermore, transient overexpression of Wnt1 or application of exogenous Wnt1 recombinant protein is necessary to preserve neurological function and rescue neurons from apoptotic membrane phosphatidylserine externalization and genomic DNA degradation, since blockade of Wnt1 signaling with a Wnt1 antibody or dickkopf related protein 1 abrogates neuronal protection by Wnt1. Wnt1 ultimately relies upon the activation of Akt1, the modulation of mitochondrial membrane permeability, and the release of cytochrome c to control the apoptotic cascade, since inhibition of Wnt1 signaling, the phosphatidylinositol 3-kinase pathway, or Akt1 activity abrogates the ability of Wnt1 to block these apoptotic components. Our work identifies Wnt1 and its downstream signaling as cellular targets with high clinical potential for novel treatment strategies for multiple disorders precipitated by oxidative stress.
Figures
Similar articles
-
EPO relies upon novel signaling of Wnt1 that requires Akt1, FoxO3a, GSK-3β, and β-catenin to foster vascular integrity during experimental diabetes.Curr Neurovasc Res. 2011 May;8(2):103-20. doi: 10.2174/156720211795495402. Curr Neurovasc Res. 2011. PMID: 21443457 Free PMC article.
-
Erythropoietin and Wnt1 govern pathways of mTOR, Apaf-1, and XIAP in inflammatory microglia.Curr Neurovasc Res. 2011 Nov;8(4):270-85. doi: 10.2174/156720211798120990. Curr Neurovasc Res. 2011. PMID: 22023617 Free PMC article.
-
Wnt1 inducible signaling pathway protein 1 (WISP1) blocks neurodegeneration through phosphoinositide 3 kinase/Akt1 and apoptotic mitochondrial signaling involving Bad, Bax, Bim, and Bcl-xL.Curr Neurovasc Res. 2012 Feb;9(1):20-31. doi: 10.2174/156720212799297137. Curr Neurovasc Res. 2012. PMID: 22272766 Free PMC article.
-
Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways.Cell Signal. 2007 Jun;19(6):1150-62. doi: 10.1016/j.cellsig.2006.12.009. Epub 2007 Jan 9. Cell Signal. 2007. PMID: 17289346 Free PMC article.
-
Erythropoietin: new directions for the nervous system.Int J Mol Sci. 2012;13(9):11102-11129. doi: 10.3390/ijms130911102. Epub 2012 Sep 6. Int J Mol Sci. 2012. PMID: 23109841 Free PMC article. Review.
Cited by
-
Antioxidants positively regulate obesity dependent circRNAs - sperm quality - functional axis.Front Endocrinol (Lausanne). 2023 Dec 11;14:1290971. doi: 10.3389/fendo.2023.1290971. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 38169845 Free PMC article.
-
Lipid Emulsion Improves Functional Recovery in an Animal Model of Stroke.Int J Mol Sci. 2020 Oct 6;21(19):7373. doi: 10.3390/ijms21197373. Int J Mol Sci. 2020. PMID: 33036206 Free PMC article.
-
Wnts are expressed in the spinal cord of adult mice and are differentially induced after injury.J Neurotrauma. 2014 Mar 15;31(6):565-81. doi: 10.1089/neu.2013.3067. J Neurotrauma. 2014. PMID: 24367909 Free PMC article.
-
Neuroprotection from Excitotoxic Injury by Local Administration of Lipid Emulsion into the Brain of Rats.Int J Mol Sci. 2020 Apr 14;21(8):2706. doi: 10.3390/ijms21082706. Int J Mol Sci. 2020. PMID: 32295117 Free PMC article.
-
Obesity and hyperglycemia lead to impaired post-ischemic recovery after permanent ischemia in mice.Obesity (Silver Spring). 2016 Feb;24(2):417-23. doi: 10.1002/oby.21388. Epub 2015 Dec 23. Obesity (Silver Spring). 2016. PMID: 26694743 Free PMC article.
References
-
- Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T, Nikolcheva T, Gotoh Y, et al. beta-Catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells. 2007;25:2827–2836. - PubMed
-
- Bakre MM, Hoi A, Mong JC, Koh YY, Wong KY, Stanton LW. Generation of multipotential mesendodermal progenitors from mouse embryonic stem cells via sustained Wnt pathway activation. J Biol Chem. 2007;282:31703–31712. - PubMed
-
- Cheng CW, Yeh JC, Fan TP, Smith SK, Charnock-Jones DS. Wnt5a-mediated non-canonical Wnt signalling regulates human endothelial cell proliferation and migration. Biochem Biophys Res Commun. 2008;365:285–290. - PubMed
-
- Jozwiak J, Kotulska K, Grajkowska W, Jozwiak S, Zalewski W, Oldak M, et al. Upregulation of the WNT pathway in tuberous sclerosis-associated subependymal giant cell astrocytomas. Brain Dev. 2007;29:273–280. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
