

Exon deletions of the EP300 and CREBBP genes in two children with Rubinstein-Taybi syndrome detected by aCGH
- PMID: 20717166
- PMCID: PMC3039495
- DOI: 10.1038/ejhg.2010.121
Exon deletions of the EP300 and CREBBP genes in two children with Rubinstein-Taybi syndrome detected by aCGH
Abstract
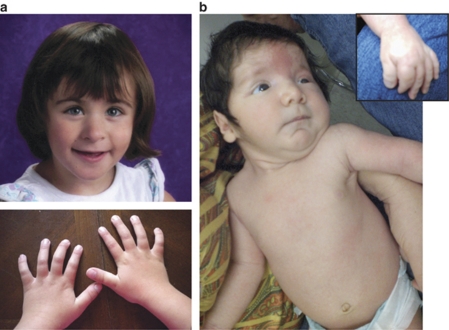
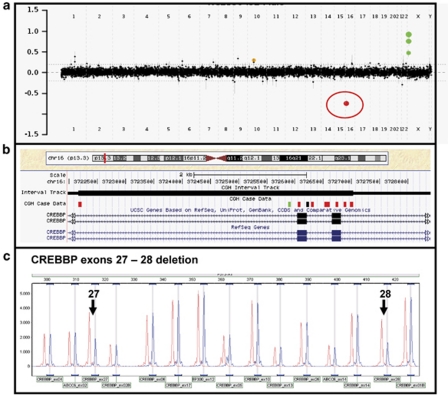
We demonstrate the utility of an exon coverage microarray platform in detecting intragenic deletions: one in exons 24-27 of the EP300 gene and another in exons 27 and 28 of the CREBBP gene in two patients with Rubinstein-Taybi syndrome (RSTS). RSTS is a heterogeneous disorder in which approximately 45-55% of cases result from deletion or mutations in the CREBBP gene and an unknown portion of cases result from gene changes in EP300. The first case is a 3-year-old female with an exonic deletion of the EP300 gene who has classic facial features of RSTS without the thumb and great toe anomalies, consistent with the milder skeletal phenotype that has been described in other RSTS cases with EP300 mutations. In addition, the mother of this patient also had preeclampsia during pregnancy, which has been infrequently reported. The second case is a newborn male who has the classical features of RSTS. Our results illustrate that exon-targeted array comparative genomic hybridization (aCGH) is a powerful tool for detecting clinically significant intragenic rearrangements that would be otherwise missed by aCGH platforms lacking sufficient exonic coverage or sequencing of the gene of interest.
Figures

References
-
- Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. 1963;105:588–608. - PubMed
-
- Petrij F, Giles RH, Dauwerse HG, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. - PubMed
-
- Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
