

Further delineation of the phenotype of severe congenital neutropenia type 4 due to mutations in G6PC3
- PMID: 20717171
- PMCID: PMC3039503
- DOI: 10.1038/ejhg.2010.136
Further delineation of the phenotype of severe congenital neutropenia type 4 due to mutations in G6PC3
Abstract
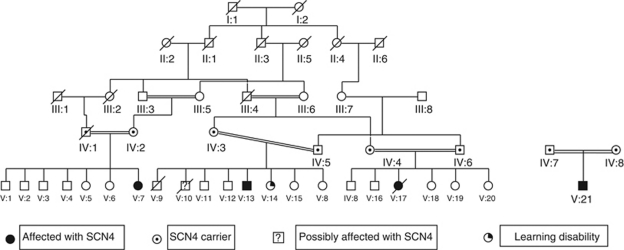
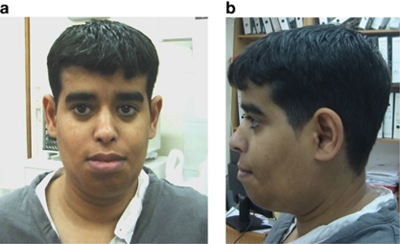
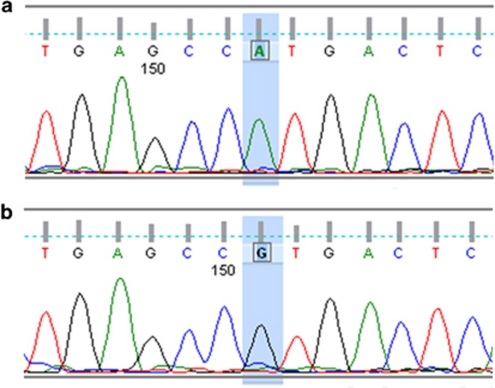
Severe congenital neutropenia type 4 (SCN4) is an autosomal recessive condition, which was defined recently with identification of the causative mutations in G6PC3. To date there are only three reports in the literature describing patients with SCN4 with mutations in the G6PC3 gene. We report four individuals with SCN4 who belong to a single large consanguineous kindred. We provide an overview of the non-haematological features of the condition with a focus on the adult phenotype, which has not been previously described in detail. We show that the superficial venous changes seen in SCN4 patients can develop into varicose veins and venous ulcers in adulthood. We review the range of congenital anomalies associated with SCN4. We demonstrate that secundum atrial septal defect, patent ductus arteriosus and valvular defects are the most frequent cardiac anomalies in SCN4. Drawing parallels with type 1 glycogen storage disease, we propose that poor growth of prenatal onset, mild-to-moderate learning disability, primary pulmonary hypertension, delayed or incomplete puberty, hypothyroidism and dysmorphism likely represent features of this syndrome. We also suggest monitoring for lipid anomalies, and kidney and liver function in affected patients. Delineation of the SCN4 phenotype may help in appropriate treatment and management and provide further insights into the pathogenesis of this multisystem disease.
Figures
Similar articles
-
Adult siblings with homozygous G6PC3 mutations expand our understanding of the severe congenital neutropenia type 4 (SCN4) phenotype.BMC Med Genet. 2012 Nov 21;13:111. doi: 10.1186/1471-2350-13-111. BMC Med Genet. 2012. PMID: 23171239 Free PMC article.
-
Severe congenital neutropenia due to G6PC3 deficiency: early and delayed phenotype in two patients with two novel mutations.Ital J Pediatr. 2014 Nov 14;40:80. doi: 10.1186/s13052-014-0080-8. Ital J Pediatr. 2014. PMID: 25391451 Free PMC article.
-
Novel G6PC3 Mutations in Patients with Congenital Neutropenia: Case Reports and Review of the Literature.Endocr Metab Immune Disord Drug Targets. 2021;21(9):1660-1668. doi: 10.2174/1871530321666210616110631. Endocr Metab Immune Disord Drug Targets. 2021. PMID: 34137364 Review.
-
A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations.Orphanet J Rare Dis. 2013 Jun 13;8:84. doi: 10.1186/1750-1172-8-84. Orphanet J Rare Dis. 2013. PMID: 23758768 Free PMC article. Review.
-
Altered Functions of Neutrophils in Two Chinese Patients With Severe Congenital Neutropenia Type 4 Caused by G6PC3 Mutations.Front Immunol. 2021 Jul 8;12:699743. doi: 10.3389/fimmu.2021.699743. eCollection 2021. Front Immunol. 2021. PMID: 34305938 Free PMC article.
Cited by
-
Drosophila neuronal Glucose-6-Phosphatase is a modulator of neuropeptide release that regulates muscle glycogen stores via FMRFamide signaling.Proc Natl Acad Sci U S A. 2024 Jul 23;121(30):e2319958121. doi: 10.1073/pnas.2319958121. Epub 2024 Jul 15. Proc Natl Acad Sci U S A. 2024. PMID: 39008673 Free PMC article.
-
Adult siblings with homozygous G6PC3 mutations expand our understanding of the severe congenital neutropenia type 4 (SCN4) phenotype.BMC Med Genet. 2012 Nov 21;13:111. doi: 10.1186/1471-2350-13-111. BMC Med Genet. 2012. PMID: 23171239 Free PMC article.
-
Hypothesis: A Novel Neuroprotective Role for Glucose-6-phosphatase (G6PC3) in Brain-To Maintain Energy-Dependent Functions Including Cognitive Processes.Neurochem Res. 2020 Nov;45(11):2529-2552. doi: 10.1007/s11064-020-03113-z. Epub 2020 Aug 19. Neurochem Res. 2020. PMID: 32815045 Review.
-
Severe congenital neutropenia due to G6PC3 deficiency: early and delayed phenotype in two patients with two novel mutations.Ital J Pediatr. 2014 Nov 14;40:80. doi: 10.1186/s13052-014-0080-8. Ital J Pediatr. 2014. PMID: 25391451 Free PMC article.
-
Family studies of warts, hypogammaglobulinemia, immunodeficiency, myelokathexis syndrome.Curr Opin Hematol. 2020 Jan;27(1):11-17. doi: 10.1097/MOH.0000000000000554. Curr Opin Hematol. 2020. PMID: 31652152 Free PMC article.
References
-
- Arostegui JI, de Toledo JS, Pascal M, Garcia C, Yague J, Diaz de Heredia C. A novel G6PC3 homozygous 1-bp deletion as a cause of severe congenital neutropenia. Blood. 2009;114:1718–1719. - PubMed
-
- Arnaiz-Villena A, Karin M, Bendikuze N, et al. HLA alleles and haplotypes in the Turkish population: relatedness to Kurds, Armenians and other Mediterraneans. Tissue Antigens. 2001;57:308–317. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
