

The crosstalk of RAS with the TGF-β family during carcinoma progression and its implications for targeted cancer therapy
- PMID: 20718708
- PMCID: PMC3044462
- DOI: 10.2174/156800910793357943
The crosstalk of RAS with the TGF-β family during carcinoma progression and its implications for targeted cancer therapy
Abstract
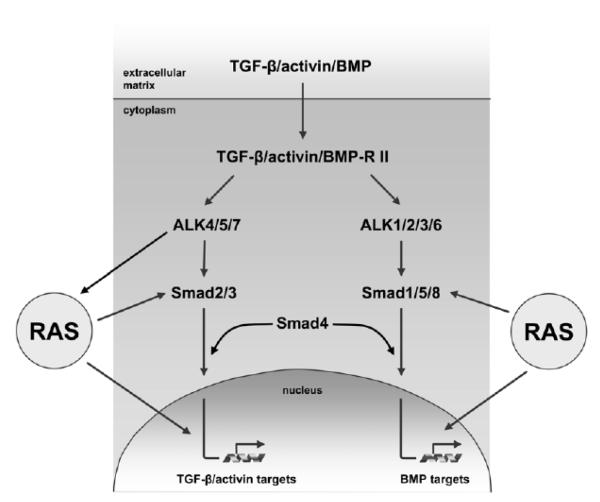
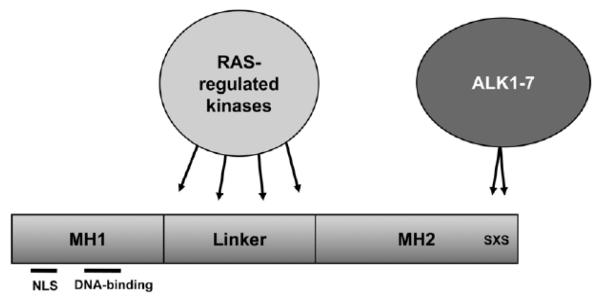
Both RAS and transforming growth factor (TGF)-β signaling cascades are central in tumorigenesis and show synergisms depending on tumor stage and tissue context. In this review we focus on the interaction of RAS subeffector proteins with signaling components of the TGF-β family including those of TGF-βs, activins and bone morphogenic proteins. Compelling evidence indicates that RAS signaling is essentially involved in the switch from tumor-suppressive to tumor-promoting functions of the TGF-β family leading to enhanced cancer growth and metastatic dissemination of primary tumors. Thus, the interface of these signaling cascades is considered as a promising target for the development of novel cancer therapeutics. The current pharmacological anti-cancer concepts combating the molecular cooperation between RAS and TGF-β family signaling during carcinoma progression are critically discussed.
Figures
Similar articles
-
TGF-β signaling in cancer.Acta Biochim Biophys Sin (Shanghai). 2018 Oct 1;50(10):941-949. doi: 10.1093/abbs/gmy092. Acta Biochim Biophys Sin (Shanghai). 2018. PMID: 30165534
-
Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis.Carcinogenesis. 2005 May;26(5):931-42. doi: 10.1093/carcin/bgi043. Epub 2005 Feb 10. Carcinogenesis. 2005. PMID: 15705598
-
TGF-β Signaling in Cancer.J Cell Biochem. 2016 Jun;117(6):1279-87. doi: 10.1002/jcb.25496. Epub 2016 Feb 11. J Cell Biochem. 2016. PMID: 26774024
-
TGF-β signaling in cancer metastasis.Acta Biochim Biophys Sin (Shanghai). 2018 Jan 1;50(1):121-132. doi: 10.1093/abbs/gmx123. Acta Biochim Biophys Sin (Shanghai). 2018. PMID: 29190313 Review.
-
Targeting the transforming growth factor-beta signaling pathway in human cancer.Expert Opin Investig Drugs. 2010 Jan;19(1):77-91. doi: 10.1517/13543780903382609. Expert Opin Investig Drugs. 2010. PMID: 20001556 Free PMC article. Review.
Cited by
-
Melatonin and TGF-β-Mediated Release of Extracellular Vesicles.Metabolites. 2023 Apr 18;13(4):575. doi: 10.3390/metabo13040575. Metabolites. 2023. PMID: 37110233 Free PMC article. Review.
-
Collagen production and niche engineering: A novel strategy for cancer cells to survive acidosis in DCIS and evolve.Evol Appl. 2020 Nov 4;13(10):2689-2703. doi: 10.1111/eva.13075. eCollection 2020 Dec. Evol Appl. 2020. PMID: 33294017 Free PMC article.
-
Genetic status of KRAS influences Transforming Growth Factor-beta (TGF-β) signaling: An insight into Neuropilin-1 (NRP1) mediated tumorigenesis.Semin Cancer Biol. 2019 Feb;54:72-79. doi: 10.1016/j.semcancer.2018.01.014. Epub 2018 Feb 2. Semin Cancer Biol. 2019. PMID: 29409705 Free PMC article. Review.
-
A microRNA code for prostate cancer metastasis.Oncogene. 2016 Mar 3;35(9):1180-92. doi: 10.1038/onc.2015.176. Epub 2015 Jun 15. Oncogene. 2016. PMID: 26073083 Free PMC article.
-
TGFβ1 regulates HRas-mediated activation of IRE1α through the PERK-RPAP2 axis in keratinocytes.Mol Carcinog. 2022 Oct;61(10):958-971. doi: 10.1002/mc.23453. Epub 2022 Aug 17. Mol Carcinog. 2022. PMID: 35975910 Free PMC article.
References
-
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer. 2003;3:362–374. - PubMed
-
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Mantovani A. Cancer: Inflaming metastasis. Nature. 2009;457:36–37. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
