

Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome
- PMID: 20720104
- PMCID: PMC2929244
- DOI: 10.1523/JNEUROSCI.1077-10.2010
Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome
Abstract
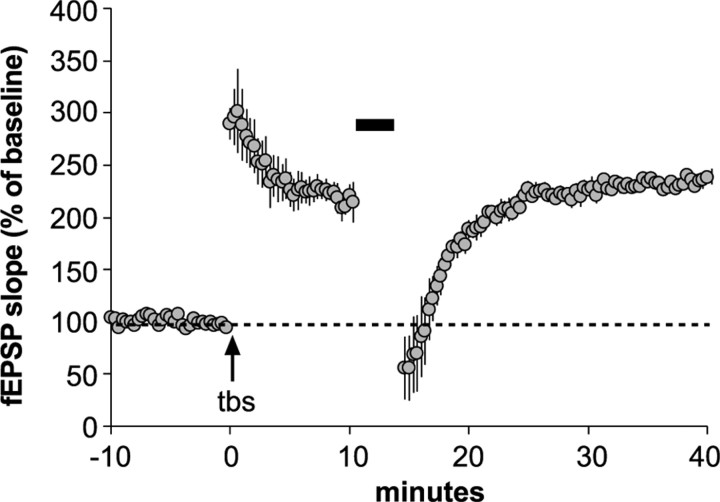
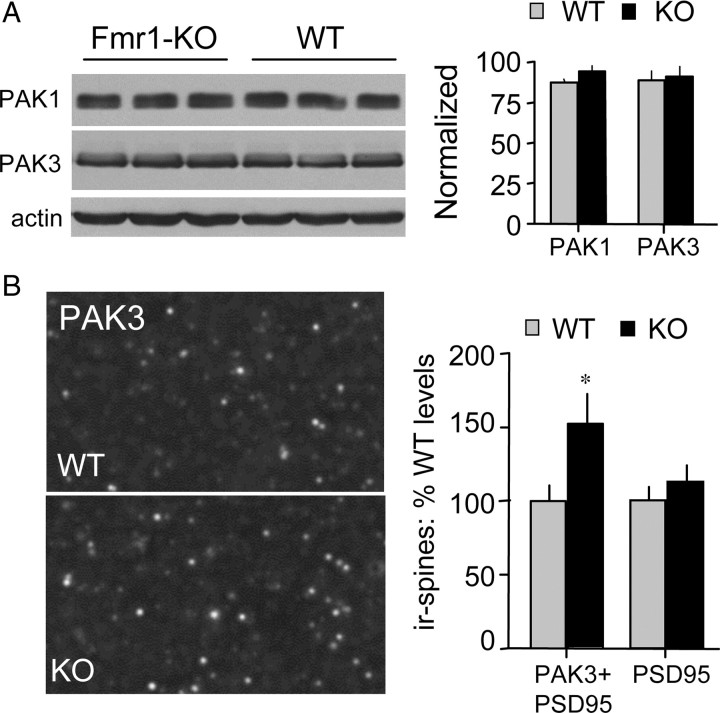
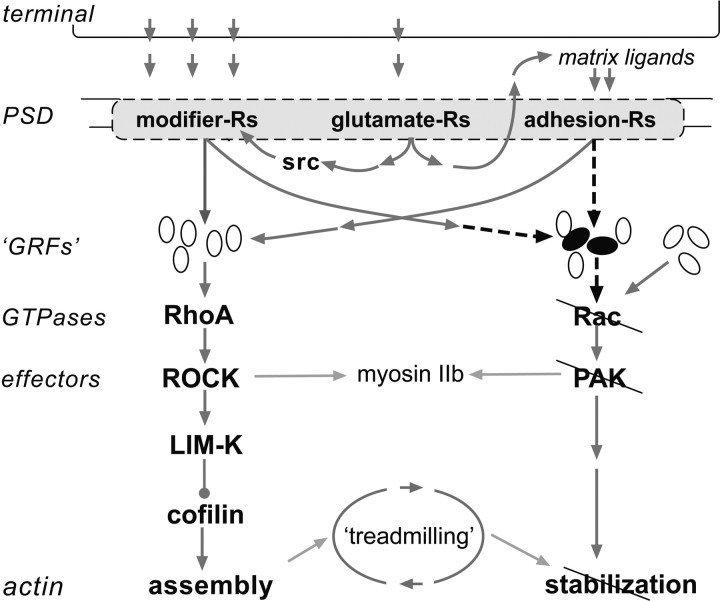
The abnormal spine morphology found in fragile X syndrome (FXS) is suggestive of an error in the signaling cascades that organize the actin cytoskeleton. We report here that physiological activation of the small GTPase Rac1 and its effector p-21 activated kinase (PAK), two enzymes critically involved in actin management and functional synaptic plasticity, is impaired at hippocampal synapses in the Fmr1-knock-out (KO) mouse model of FXS. Theta burst afferent stimulation (TBS) caused a marked increase in the number of synapses associated with phosphorylated PAK in adult hippocampal slices from wild-type, but not Fmr1-KO, mice. Stimulation-induced activation of synaptic Rac1 was also absent in the mutants. The polymerization of spine actin that occurs immediately after theta stimulation appeared normal in mutant slices but the newly formed polymers did not properly stabilize, as evidenced by a prolonged vulnerability to a toxin (latrunculin) that disrupts dynamic actin filaments. Latrunculin also reversed long-term potentiation when applied at 10 min post-TBS, a time point at which the potentiation effect is resistant to interference in wild-type slices. We propose that a Rac>PAK signaling pathway needed for rapid stabilization of activity-induced actin filaments, and thus for normal spine morphology and lasting synaptic changes, is defective in FXS.
Figures



Similar articles
-
Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome.Sci Signal. 2017 Nov 7;10(504):eaan0852. doi: 10.1126/scisignal.aan0852. Sci Signal. 2017. PMID: 29114038 Free PMC article.
-
Increased Training Intensity Induces Proper Membrane Localization of Actin Remodeling Proteins in the Hippocampus Preventing Cognitive Deficits: Implications for Fragile X Syndrome.Mol Neurobiol. 2018 Jun;55(6):4529-4542. doi: 10.1007/s12035-017-0666-4. Epub 2017 Jul 8. Mol Neurobiol. 2018. PMID: 28688003
-
Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome.J Neurosci. 2007 Oct 3;27(40):10685-94. doi: 10.1523/JNEUROSCI.2624-07.2007. J Neurosci. 2007. PMID: 17913902 Free PMC article.
-
Imbalance of synaptic actin dynamics as a key to fragile X syndrome?J Physiol. 2018 Jul;596(14):2773-2782. doi: 10.1113/JP275571. Epub 2018 Feb 25. J Physiol. 2018. PMID: 29380377 Free PMC article. Review.
-
Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity?Neuroscientist. 2012 Feb;18(1):28-44. doi: 10.1177/1073858410395322. Epub 2011 May 6. Neuroscientist. 2012. PMID: 21551076 Review.
Cited by
-
Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486.Proc Natl Acad Sci U S A. 2013 Apr 2;110(14):5671-6. doi: 10.1073/pnas.1219383110. Epub 2013 Mar 18. Proc Natl Acad Sci U S A. 2013. PMID: 23509247 Free PMC article.
-
Integrin dynamics produce a delayed stage of long-term potentiation and memory consolidation.J Neurosci. 2012 Sep 12;32(37):12854-61. doi: 10.1523/JNEUROSCI.2024-12.2012. J Neurosci. 2012. PMID: 22973009 Free PMC article.
-
Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines.Cytoskeleton (Hoboken). 2012 Jul;69(7):426-41. doi: 10.1002/cm.21015. Epub 2012 Mar 12. Cytoskeleton (Hoboken). 2012. PMID: 22307832 Free PMC article. Review.
-
Inka2, a novel Pak4 inhibitor, regulates actin dynamics in neuronal development.PLoS Genet. 2022 Oct 27;18(10):e1010438. doi: 10.1371/journal.pgen.1010438. eCollection 2022 Oct. PLoS Genet. 2022. PMID: 36301793 Free PMC article.
-
PAK family kinases: Physiological roles and regulation.Cell Logist. 2012 Apr 1;2(2):59-68. doi: 10.4161/cl.21912. Cell Logist. 2012. PMID: 23162738 Free PMC article.
References
-
- Bardoni B, Mandel JL. Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr Opin Genet Dev. 2002;12:284–293. - PubMed
-
- Billuart P, Chelly J. From fragile X mental retardation protein to Rac1 GTPase: new insights from Fly CYFIP. Neuron. 2003;38:843–845. - PubMed
-
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous