

Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease
- PMID: 20725711
- PMCID: PMC4319704
- DOI: 10.1007/s00109-010-0663-9
Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease
Abstract
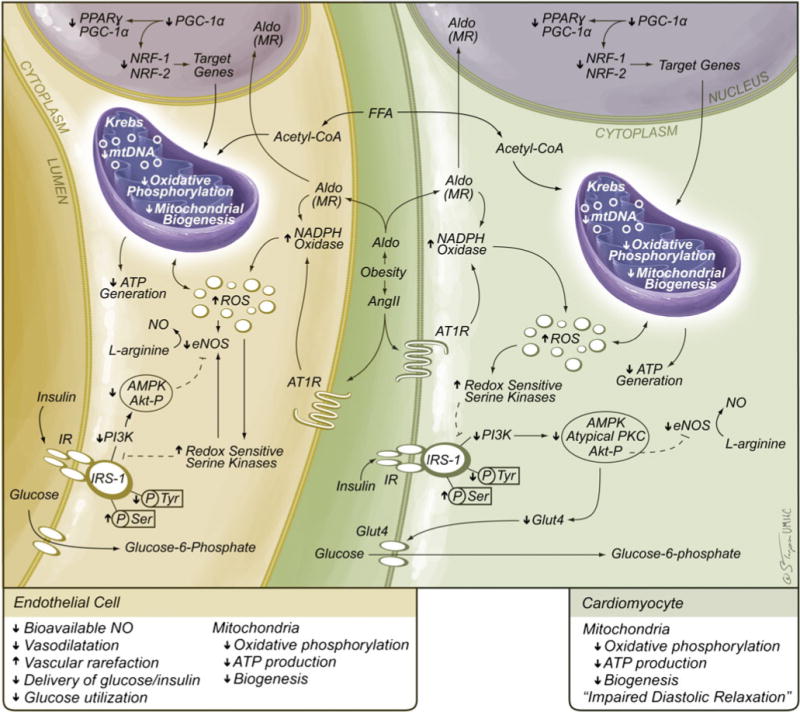
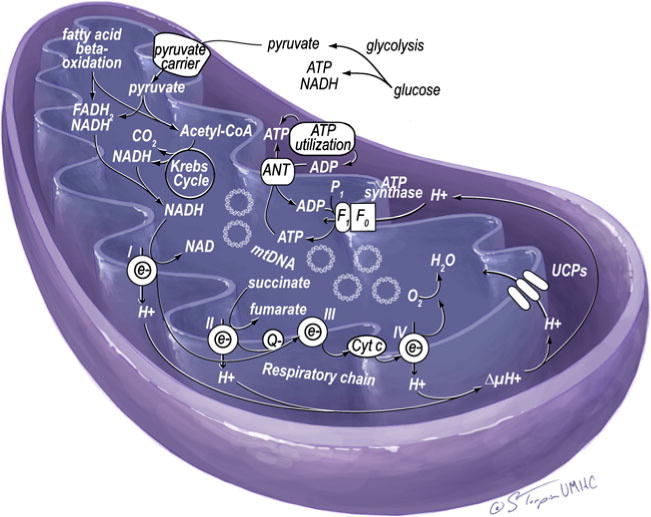
The metabolic syndrome is a constellation of metabolic disorders including obesity, hypertension, and insulin resistance, components which are risk factors for the development of diabetes, hypertension, cardiovascular, and renal disease. Pathophysiological abnormalities that contribute to the development of the metabolic syndrome include impaired mitochondrial oxidative phosphorylation and mitochondrial biogenesis, dampened insulin metabolic signaling, endothelial dysfunction, and associated myocardial functional abnormalities. Recent evidence suggests that impaired myocardial mitochondrial biogenesis, fatty acid metabolism, and antioxidant defense mechanisms lead to diminished cardiac substrate flexibility, decreased cardiac energetic efficiency, and diastolic dysfunction. In addition, enhanced activation of the renin-angiotensin-aldosterone system and associated increases in oxidative stress can lead to mitochondrial apoptosis and degradation, altered bioenergetics, and accumulation of lipids in the heart. In addition to impairments in metabolic signaling and oxidative stress, genetic and environmental factors, aging, and hyperglycemia all contribute to reduced mitochondrial biogenesis and mitochondrial dysfunction. These mitochondrial abnormalities can predispose a metabolic cardiomyopathy characterized by diastolic dysfunction. Mitochondrial dysfunction and resulting lipid accumulation in skeletal muscle, liver, and pancreas also impede insulin metabolic signaling and glucose metabolism, ultimately leading to a further increase in mitochondrial dysfunction. Interventions to improve mitochondrial function have been shown to correct insulin metabolic signaling and other metabolic and cardiovascular abnormalities. This review explores mechanisms of mitochondrial dysfunction with a focus on impaired oxidative phosphorylation and mitochondrial biogenesis in the pathophysiology of metabolic heart disease.
Conflict of interest statement
Figures

References
-
- Sullivan PW, Ghushchyan V, Wyatt HR, Wu EQ, Hill JO. Impact of cardiometabolic risk factor clusters on health-related quality of life in the U.S. Obesity (Silver Spring) 2007;15:511–521. - PubMed
-
- Cannon CP, Kumar A. Treatment of overweight and obesity: lifestyle, pharmacologic, and surgical options. Clin Cornerstone. 2009;9:55–68. discussion 69–71. - PubMed
-
- Sowers JR. Metabolic risk factors and renal disease. Kidney Int. 2007;71:719–720. - PubMed
-
- Smith SC., Jr Multiple risk factors for cardiovascular disease and diabetes mellitus. Am J Med. 2007;120:S3–S11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
