

Introducing knowledge into differential expression analysis
- PMID: 20726790
- PMCID: PMC3122906
- DOI: 10.1089/cmb.2010.0034
Introducing knowledge into differential expression analysis
Abstract
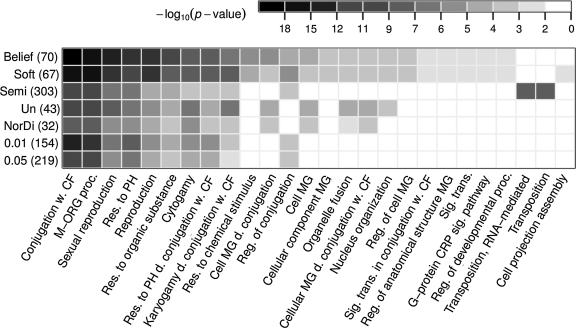
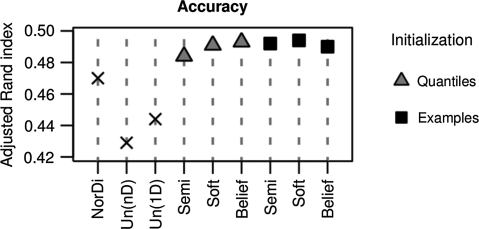
Gene expression measurements allow determining sets of up- or down-regulated, or unchanged genes in a particular experimental condition. Additional biological knowledge can suggest examples of genes from one of these sets. For instance, known target genes of a transcriptional activator are expected, but are not certain to go down after this activator is knocked out. Available differential expression analysis tools do not take such imprecise examples into account. Here we put forward a novel partially supervised mixture modeling methodology for differential expression analysis. Our approach, guided by imprecise examples, clusters expression data into differentially expressed and unchanged genes. The partially supervised methodology is implemented by two methods: a newly introduced belief-based mixture modeling, and soft-label mixture modeling, a method proved efficient in other applications. We investigate on synthetic data the input example settings favorable for each method. In our tests, both belief-based and soft-label methods prove their advantage over semi-supervised mixture modeling in correcting for erroneous examples. We also compare them to alternative differential expression analysis approaches, showing that incorporation of knowledge yields better performance. We present a broad range of knowledge sources and data to which our partially supervised methodology can be applied. First, we determine targets of Ste12 based on yeast knockout data, guided by a Ste12 DNA-binding experiment. Second, we distinguish miR-1 from miR-124 targets in human by clustering expression data under transfection experiments of both microRNAs, using their computationally predicted targets as examples. Finally, we utilize literature knowledge to improve clustering of time-course expression profiles.
Figures





Similar articles
-
Beyond synexpression relationships: local clustering of time-shifted and inverted gene expression profiles identifies new, biologically relevant interactions.J Mol Biol. 2001 Dec 14;314(5):1053-66. doi: 10.1006/jmbi.2000.5219. J Mol Biol. 2001. PMID: 11743722
-
The TEA transcription factor Tec1 confers promoter-specific gene regulation by Ste12-dependent and -independent mechanisms.Eukaryot Cell. 2010 Apr;9(4):514-31. doi: 10.1128/EC.00251-09. Epub 2010 Jan 29. Eukaryot Cell. 2010. PMID: 20118212 Free PMC article.
-
Co-clustering and visualization of gene expression data and gene ontology terms for Saccharomyces cerevisiae using self-organizing maps.J Biomed Inform. 2007 Apr;40(2):160-73. doi: 10.1016/j.jbi.2006.05.001. Epub 2006 May 20. J Biomed Inform. 2007. PMID: 16824804
-
Transcriptional networks: reverse-engineering gene regulation on a global scale.Curr Opin Microbiol. 2004 Dec;7(6):638-46. doi: 10.1016/j.mib.2004.10.009. Curr Opin Microbiol. 2004. PMID: 15556037 Review.
-
Ste12 and Ste12-like proteins, fungal transcription factors regulating development and pathogenicity.Eukaryot Cell. 2010 Apr;9(4):480-5. doi: 10.1128/EC.00333-09. Epub 2010 Feb 5. Eukaryot Cell. 2010. PMID: 20139240 Free PMC article. Review.
Cited by
-
PROmiRNA: a new miRNA promoter recognition method uncovers the complex regulation of intronic miRNAs.Genome Biol. 2013 Aug 16;14(8):R84. doi: 10.1186/gb-2013-14-8-r84. Genome Biol. 2013. PMID: 23958307 Free PMC article.
-
Deregulation upon DNA damage revealed by joint analysis of context-specific perturbation data.BMC Bioinformatics. 2011 Jun 21;12:249. doi: 10.1186/1471-2105-12-249. BMC Bioinformatics. 2011. PMID: 21693013 Free PMC article.
-
Inhibition decorrelates visual feature representations in the inner retina.Nature. 2017 Feb 23;542(7642):439-444. doi: 10.1038/nature21394. Epub 2017 Feb 8. Nature. 2017. PMID: 28178238 Free PMC article.
References
-
- Alexandridis R. Lin S. Irwin M. Class discovery and classification of tumor samples using mixture modeling of gene expression data—a unified approach. Bioinformatics. 2004;20:2545–2552. - PubMed
-
- Baldi P. Long A.D. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. - PubMed
-
- Betel D. Wilson M. Gabow A., et al. The microRNA.org. resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases