

A haplotype inference algorithm for trios based on deterministic sampling
- PMID: 20727218
- PMCID: PMC2939632
- DOI: 10.1186/1471-2156-11-78
A haplotype inference algorithm for trios based on deterministic sampling
Abstract
Background: In genome-wide association studies, thousands of individuals are genotyped in hundreds of thousands of single nucleotide polymorphisms (SNPs). Statistical power can be increased when haplotypes, rather than three-valued genotypes, are used in analysis, so the problem of haplotype phase inference (phasing) is particularly relevant. Several phasing algorithms have been developed for data from unrelated individuals, based on different models, some of which have been extended to father-mother-child "trio" data.
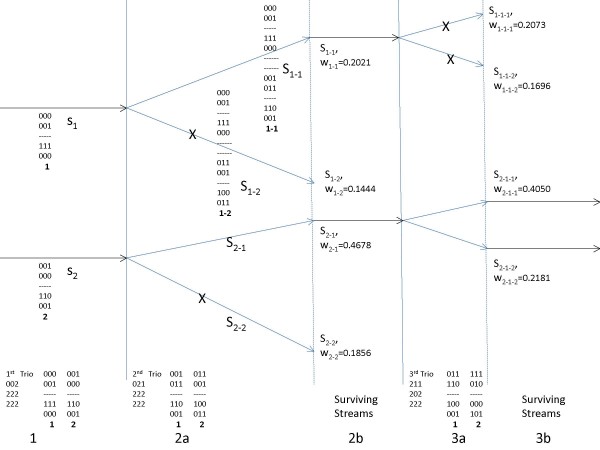
Results: We introduce a technique for phasing trio datasets using a tree-based deterministic sampling scheme. We have compared our method with publicly available algorithms PHASE v2.1, BEAGLE v3.0.2 and 2SNP v1.7 on datasets of varying number of markers and trios. We have found that the computational complexity of PHASE makes it prohibitive for routine use; on the other hand 2SNP, though the fastest method for small datasets, was significantly inaccurate. We have shown that our method outperforms BEAGLE in terms of speed and accuracy for small to intermediate dataset sizes in terms of number of trios for all marker sizes examined. Our method is implemented in the "Tree-Based Deterministic Sampling" (TDS) package, available for download at http://www.ee.columbia.edu/~anastas/tds
Conclusions: Using a Tree-Based Deterministic sampling technique, we present an intuitive and conceptually simple phasing algorithm for trio data. The trade off between speed and accuracy achieved by our algorithm makes it a strong candidate for routine use on trio datasets.
Figures
Similar articles
-
Fast and accurate haplotype frequency estimation for large haplotype vectors from pooled DNA data.BMC Genet. 2012 Oct 30;13:94. doi: 10.1186/1471-2156-13-94. BMC Genet. 2012. PMID: 23110720 Free PMC article.
-
A unified framework for haplotype inference in nuclear families.Ann Hum Genet. 2012 Jul;76(4):312-25. doi: 10.1111/j.1469-1809.2012.00715.x. Epub 2012 May 21. Ann Hum Genet. 2012. PMID: 22607042
-
2SNP: scalable phasing method for trios and unrelated individuals.IEEE/ACM Trans Comput Biol Bioinform. 2008 Apr-Jun;5(2):313-8. doi: 10.1109/TCBB.2007.1068. IEEE/ACM Trans Comput Biol Bioinform. 2008. PMID: 18451440
-
Missing data imputation and haplotype phase inference for genome-wide association studies.Hum Genet. 2008 Dec;124(5):439-50. doi: 10.1007/s00439-008-0568-7. Epub 2008 Oct 11. Hum Genet. 2008. PMID: 18850115 Free PMC article. Review.
-
A Pipeline for Phasing and Genotype Imputation on Mixed Human Data (Parents-Offspring Trios and Unrelated Subjects) by Reviewing Current Methods and Software.Life (Basel). 2022 Dec 5;12(12):2030. doi: 10.3390/life12122030. Life (Basel). 2022. PMID: 36556394 Free PMC article. Review.
Cited by
-
Curiosities of X chromosomal markers and haplotypes.Int J Legal Med. 2018 Mar;132(2):361-371. doi: 10.1007/s00414-017-1612-8. Epub 2017 May 26. Int J Legal Med. 2018. PMID: 28547136
-
A sequential Monte Carlo framework for haplotype inference in CNV/SNP genotype data.EURASIP J Bioinform Syst Biol. 2014;2014(1):7. doi: 10.1186/1687-4153-2014-7. Epub 2014 Apr 24. EURASIP J Bioinform Syst Biol. 2014. PMID: 24868199 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
