

Defining new insight into atypical arrhythmia: a computational model of ankyrin-B syndrome
- PMID: 20729400
- PMCID: PMC2993217
- DOI: 10.1152/ajpheart.00503.2010
Defining new insight into atypical arrhythmia: a computational model of ankyrin-B syndrome
Abstract
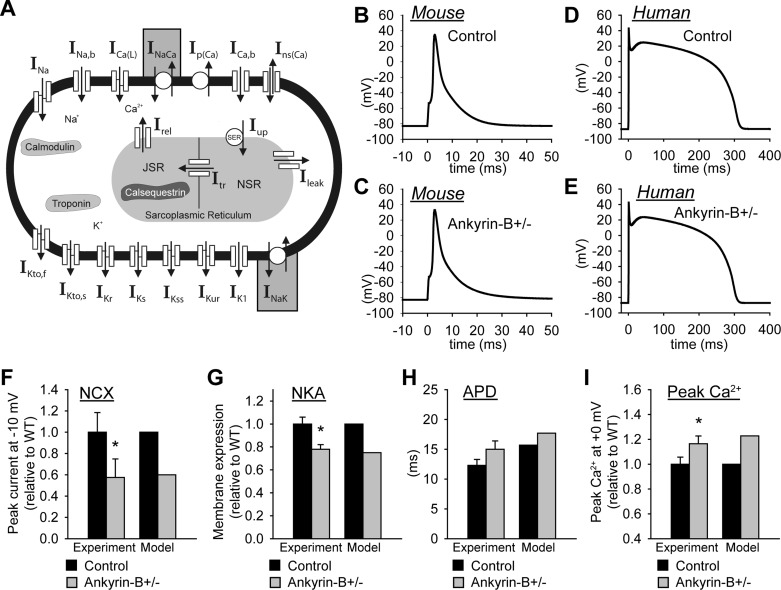
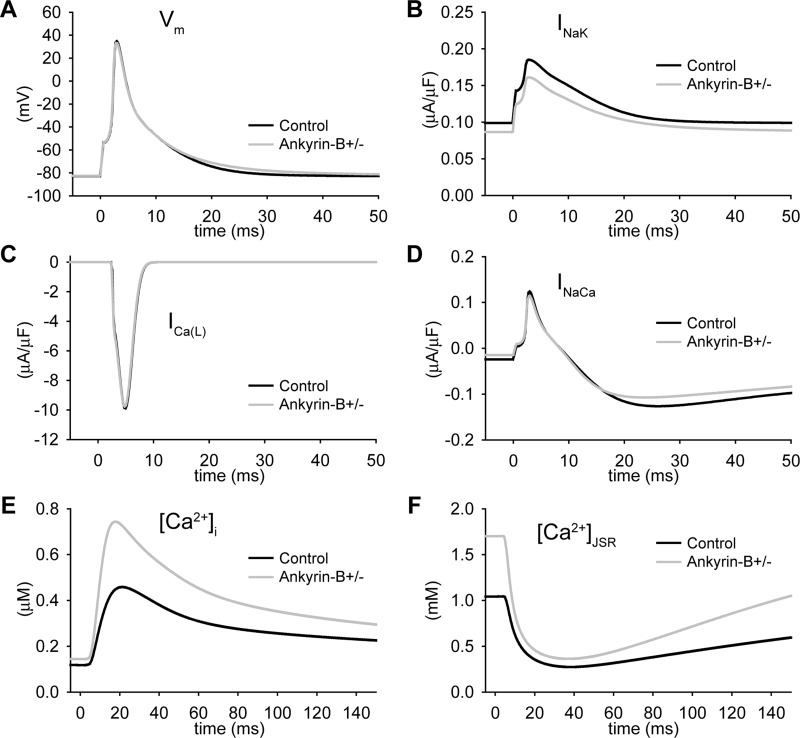
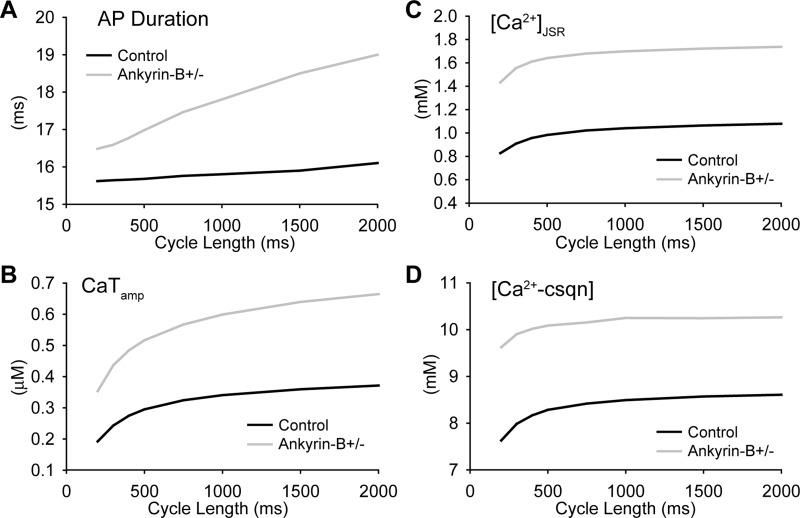
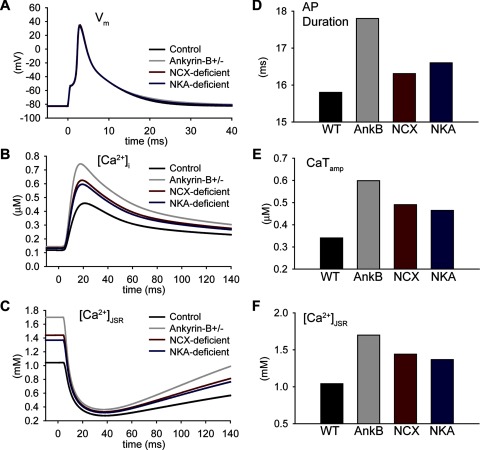
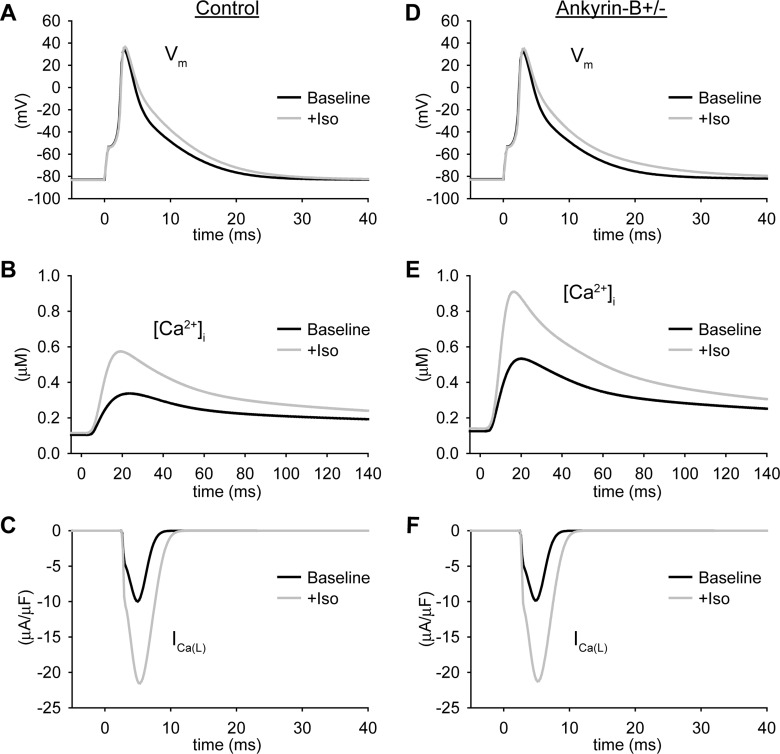
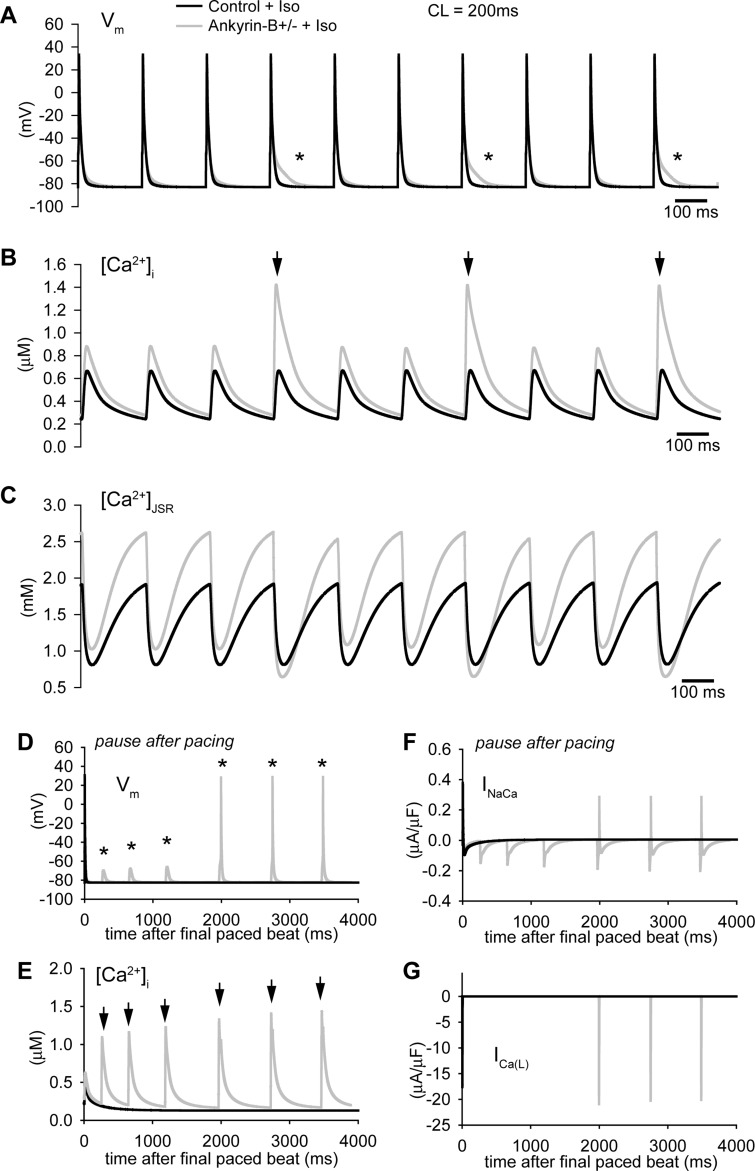
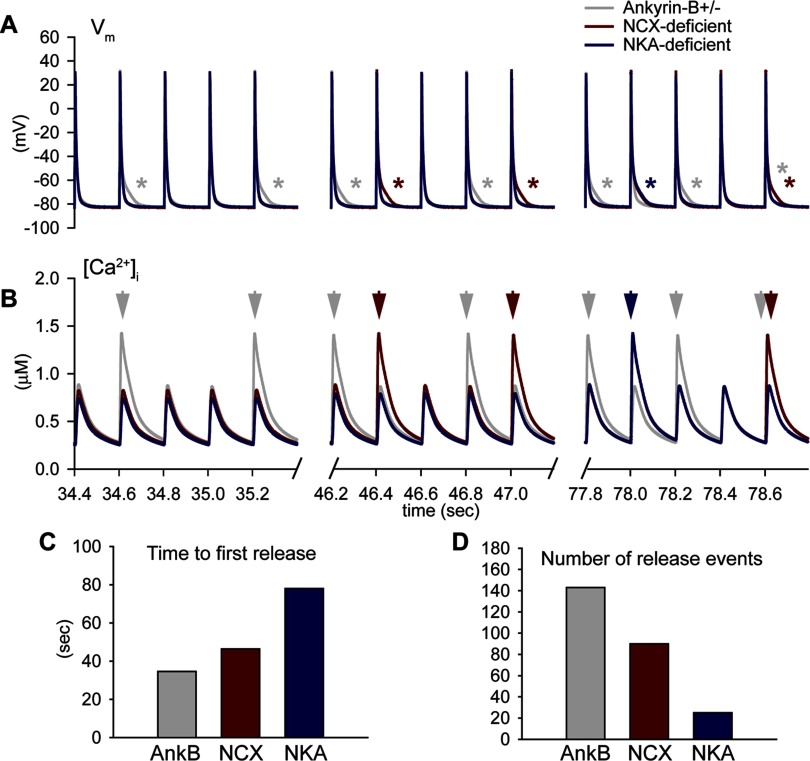
Normal cardiac excitability depends on the coordinated activity of specific ion channels and transporters within specialized domains at the plasma membrane and sarcoplasmic reticulum. Ion channel dysfunction due to congenital or acquired defects has been linked to human cardiac arrhythmia. More recently, defects in ion channel-associated proteins have been associated with arrhythmia. Ankyrin-B is a multifunctional adapter protein responsible for targeting select ion channels, transporters, cytoskeletal proteins, and signaling molecules in excitable cells, including neurons, pancreatic β-cells, and cardiomyocytes. Ankyrin-B dysfunction has been linked to cardiac arrhythmia in human patients and ankyrin-B heterozygous (ankyrin-B(+/-)) mice with a phenotype characterized by sinus node dysfunction, susceptibility to ventricular arrhythmias, and sudden death ("ankyrin-B syndrome"). At the cellular level, ankyrin-B(+/-) cells have defects in the expression and membrane localization of the Na(+)/Ca(2+) exchanger and Na(+)-K(+)-ATPase, Ca(2+) overload, and frequent afterdepolarizations, which likely serve as triggers for lethal cardiac arrhythmias. Despite knowledge gathered from mouse models and human patients, the molecular mechanism responsible for cardiac arrhythmias in the setting of ankyrin-B dysfunction remains unclear. Here, we use mathematical modeling to provide new insights into the cellular pathways responsible for Ca(2+) overload and afterdepolarizations in ankyrin-B(+/-) cells. We show that the Na(+)/Ca(2+) exchanger and Na(+)-K(+)-ATPase play related, yet distinct, roles in intracellular Ca(2+) accumulation, sarcoplasmic reticulum Ca(2+) overload, and afterdepolarization generation in ankyrin-B(+/-) cells. These findings provide important insights into the molecular mechanisms underlying a human disease and are relevant for acquired human arrhythmia, where ankyrin-B dysfunction has recently been identified.
Figures
Similar articles
-
Atrial fibrillation and sinus node dysfunction in human ankyrin-B syndrome: a computational analysis.Am J Physiol Heart Circ Physiol. 2013 May;304(9):H1253-66. doi: 10.1152/ajpheart.00734.2012. Epub 2013 Feb 22. Am J Physiol Heart Circ Physiol. 2013. PMID: 23436330 Free PMC article.
-
A cardiac arrhythmia syndrome caused by loss of ankyrin-B function.Proc Natl Acad Sci U S A. 2004 Jun 15;101(24):9137-42. doi: 10.1073/pnas.0402546101. Epub 2004 Jun 3. Proc Natl Acad Sci U S A. 2004. PMID: 15178757 Free PMC article.
-
CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome.Heart Rhythm. 2012 Dec;9(12):2034-41. doi: 10.1016/j.hrthm.2012.08.026. Epub 2012 Aug 28. Heart Rhythm. 2012. PMID: 23059182 Free PMC article.
-
Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart.Cardiovasc Res. 2006 Jul 1;71(1):22-9. doi: 10.1016/j.cardiores.2006.03.018. Epub 2006 Mar 28. Cardiovasc Res. 2006. PMID: 16650839 Review.
-
Ankyrin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting.Curr Opin Cardiol. 2005 May;20(3):189-93. doi: 10.1097/01.hco.0000160372.95116.3e. Curr Opin Cardiol. 2005. PMID: 15861006 Review.
Cited by
-
Defects in cytoskeletal signaling pathways, arrhythmia, and sudden cardiac death.Front Physiol. 2012 May 3;3:122. doi: 10.3389/fphys.2012.00122. eCollection 2012. Front Physiol. 2012. PMID: 22586405 Free PMC article.
-
Simulating Notch-Dome Morphology of Action Potential of Ventricular Cell: How the Speeds of Positive and Negative Feedbacks on Transmembrane Voltage Can Influence the Health of a Cell?Biomed Res Int. 2020 Sep 3;2020:5169241. doi: 10.1155/2020/5169241. eCollection 2020. Biomed Res Int. 2020. PMID: 32953882 Free PMC article.
-
Ankyrins and Spectrins in Cardiovascular Biology and Disease.Front Physiol. 2017 Oct 27;8:852. doi: 10.3389/fphys.2017.00852. eCollection 2017. Front Physiol. 2017. PMID: 29163198 Free PMC article. Review.
-
Coordinating electrical activity of the heart: ankyrin polypeptides in human cardiac disease.Expert Opin Ther Targets. 2011 Jul;15(7):789-801. doi: 10.1517/14728222.2011.575363. Epub 2011 Apr 4. Expert Opin Ther Targets. 2011. PMID: 21457127 Free PMC article. Review.
-
Atrial fibrillation and sinus node dysfunction in human ankyrin-B syndrome: a computational analysis.Am J Physiol Heart Circ Physiol. 2013 May;304(9):H1253-66. doi: 10.1152/ajpheart.00734.2012. Epub 2013 Feb 22. Am J Physiol Heart Circ Physiol. 2013. PMID: 23436330 Free PMC article.
References
-
- Ayalon G, Davis JQ, Scotland PB, Bennett V. An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell 135: 1189– 1200, 2008. - PubMed
-
- Bhasin N, Cunha SR, Mduannayake M, Gigena MS, Rogers TB, Mohler PJ. Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am J Physiol Heart Circ Physiol 293: H109– H119, 2007. - PubMed
-
- Bondarenko VE, Szigeti GP, Bett GC, Kim SJ, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 287: H1378– H1403, 2004. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous