

How adaptation of the brain to alcohol leads to dependence: a pharmacological perspective
- PMID: 20729980
- PMCID: PMC2923844
How adaptation of the brain to alcohol leads to dependence: a pharmacological perspective
Abstract
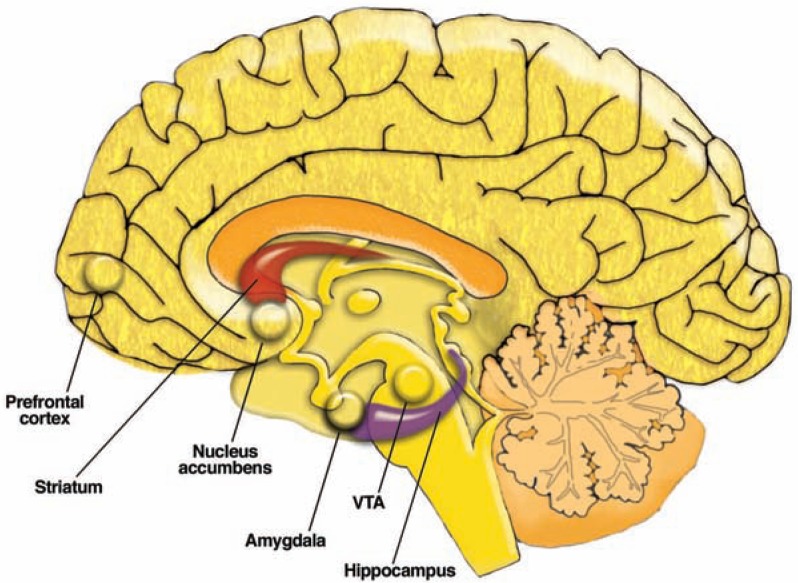
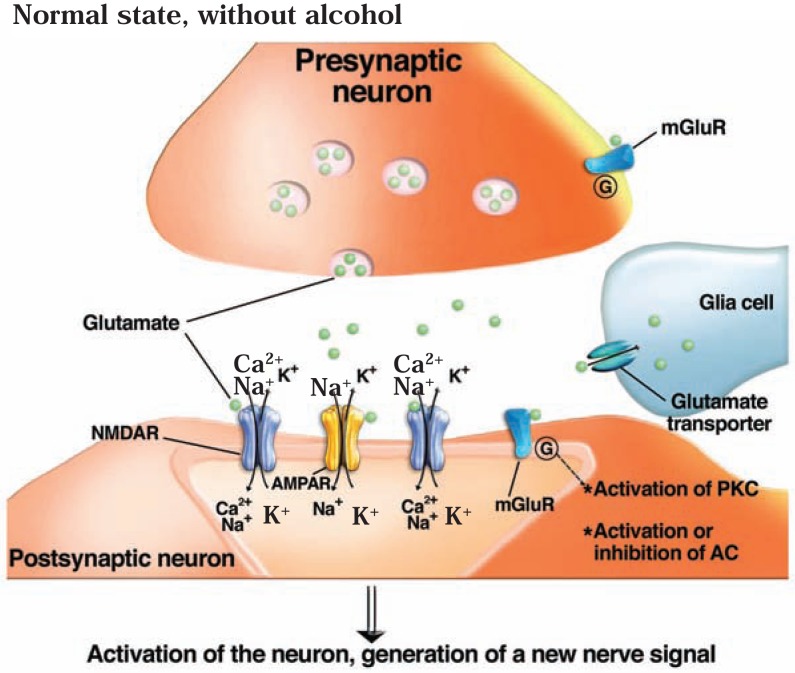
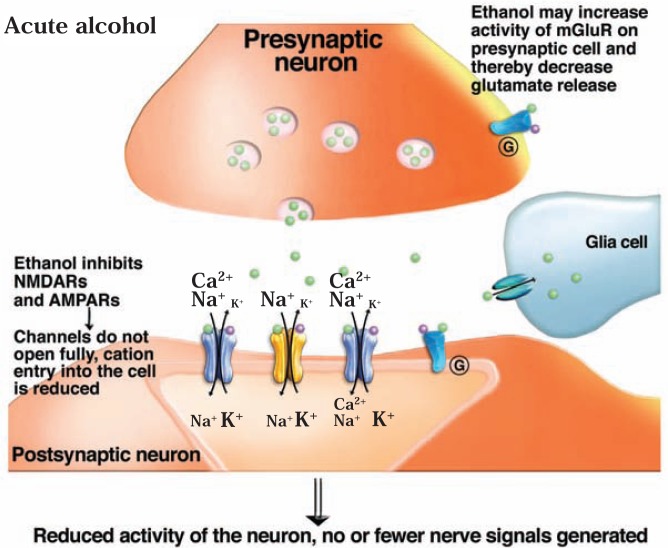
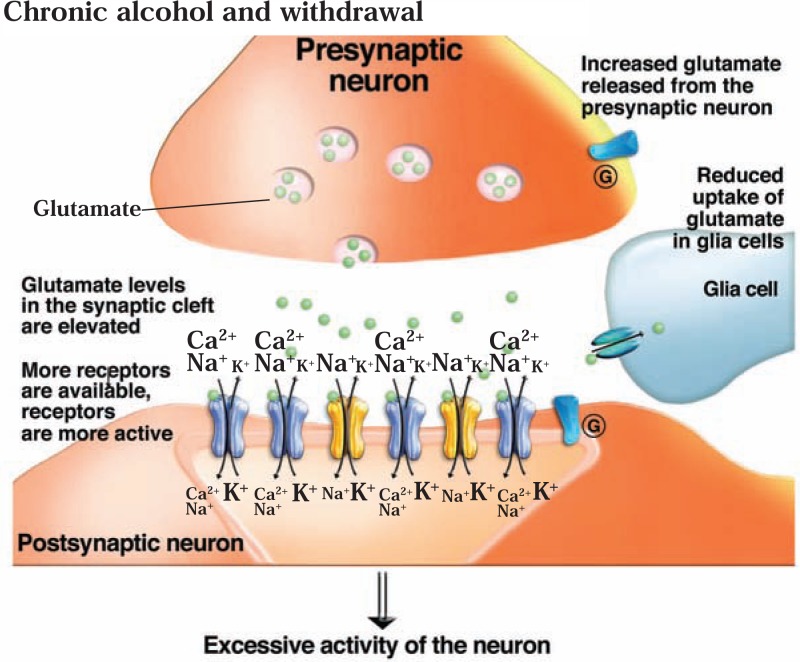
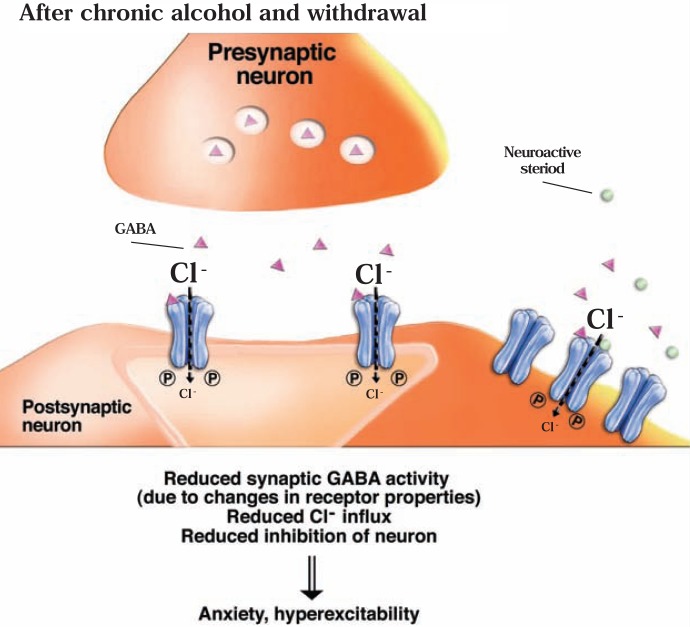
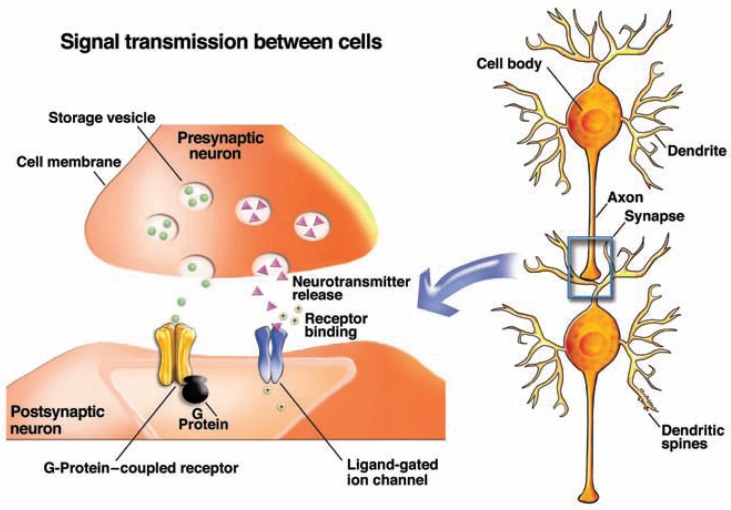
The development of alcohol dependence is posited to involve numerous changes in brain chemistry (i.e., neurotransmission) that lead to physiological signs of withdrawal upon abstinence from alcohol as well as promote vulnerability to relapse in dependent people. These neuroadaptive changes often occur in those brain neurotransmission systems that are most sensitive to the acute, initial effects of alcohol and/or contribute to a person’s initial alcohol consumption. Studies of these neuroadaptive changes have been aided by the development of animal models of alcohol dependence, withdrawal, and relapse behavior. These animal models, as well as findings obtained in humans, have shed light on the effects that acute and chronic alcohol exposure have on signaling systems involving the neurotransmitters glutamate, γaminobutyric acid (GABA), dopamine, and serotonin, as well as on other signaling molecules, including endogenous opioids and corticotrophin-releasing factor (CRF). Adaptation to chronic alcohol exposure by these systems has been associated with behavioral effects, such as changes in reinforcement, enhanced anxiety, and increased sensitivity to stress, all of which may contribute to relapse to drinking in abstinent alcoholics. Moreover, some of these systems are targets of currently available therapeutic agents for alcohol dependence.
Figures
References
-
- Ait-Daoud N, Johnson BA, Javors M, et al. Combining ondansetron and naltrexone treats biological alcoholics: Corroboration of self-reported drinking by serum carbohydrate deficient transferrin, a biomarker. Alcoholism: Clinical and Experimental Research. 2001;25:847–849. - PubMed
-
- Boyce-Rustay JM, Wiedholz LM, Millstein RA, et al. Ethanol-related behaviors in serotonin transporter knockout mice. Alcoholism: Clinical and Experimental Research. 2006;30:1957–1965. - PubMed
-
- Casu MA, Pisu C, Lobina C, Pani L. Immunocytochemical study of the forebrain serotonergic innervation in Sardinian alcohol-preferring rats. Psychopharmacology (Berlin) 2004;172:341–351. - PubMed
-
- Colombo G, Orru A, Lai P, et al. The cannabinoid CB1 receptor antagonist, rimonabant, as a promising pharmacotherapy for alcohol dependence: Preclinical evidence. Molecular Neurobiology. 2007;36:102–112. - PubMed
-
- Hodge CW, Kelley SP, Bratt AM, et al. 5-HT(3A) receptor subunit is required for 5-HT3 antagonist-induced reductions in alcohol drinking. Neuropsychopharmacology. 2004;29:1807–1813. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical