

Chapter 4. Predicting and characterizing protein functions through matching geometric and evolutionary patterns of binding surfaces
- PMID: 20731991
- PMCID: PMC2882714
- DOI: 10.1016/S0065-3233(07)75004-0
Chapter 4. Predicting and characterizing protein functions through matching geometric and evolutionary patterns of binding surfaces
Abstract
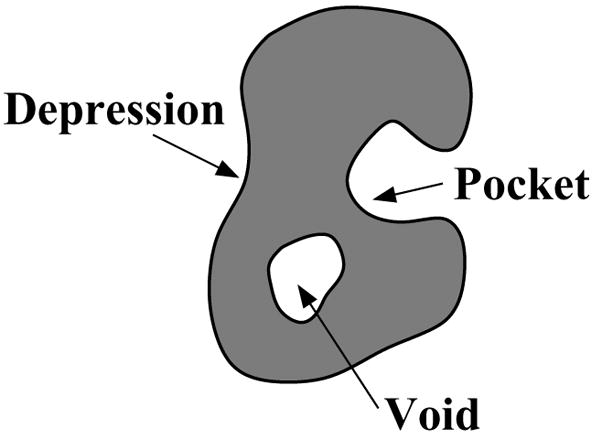
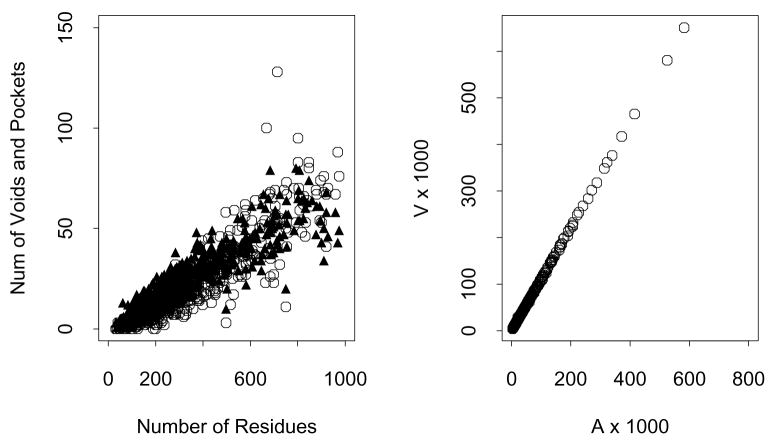
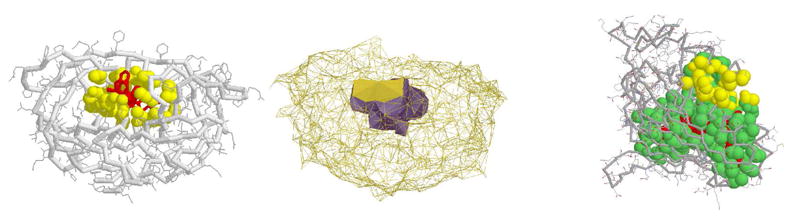
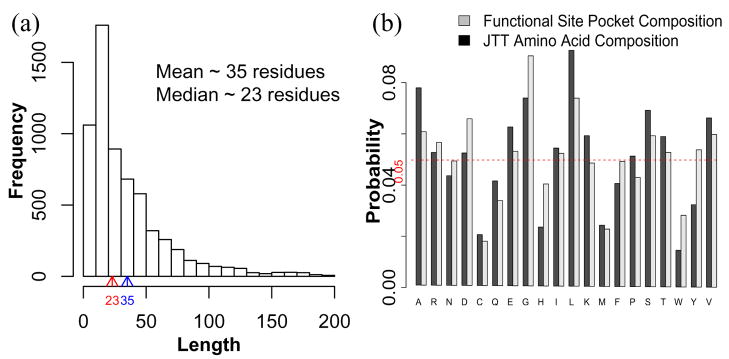
Predicting protein functions from structures is an important and challenging task. Although proteins are often thought to be packed as tightly as solids, closer examination based on geometric computation reveals that they contain numerous voids and pockets. Most of them are of random nature, but some are binding sites providing surfaces to interact with other molecules. A promising approach for function prediction is to infer functions through discovery of similarity in local binding pockets, as proteins binding to similar substrates/ligands and carrying out similar functions have similar physical constraints for binding and reactions. In this chapter, we describe computational methods to distinguish those surface pockets that are likely to be involved in important biological functions, and methods to identify key residues in these pockets. We further describe how to predict protein functions at large scale from structures by detecting binding surfaces similar in residue make-ups, shape, and orientation. We also describe a Bayesian Monte Carlo method that can separate selection pressure due to biological function from pressure due to protein folding. We show how this method can be used to reconstruct the evolutionary history of binding surfaces for detecting similar binding surfaces. In addition, we briefly discuss how the negative image of a binding pocket can be casted, and how such information can be used to facilitate drug discovery.
Keywords: Bayesian Monte Carlo; CASTp; Local binding surface; alpha shape; pocket; protein function; pvSOAR; void.
Figures
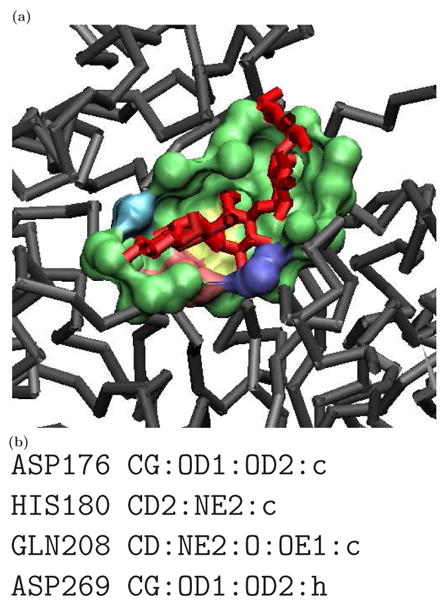
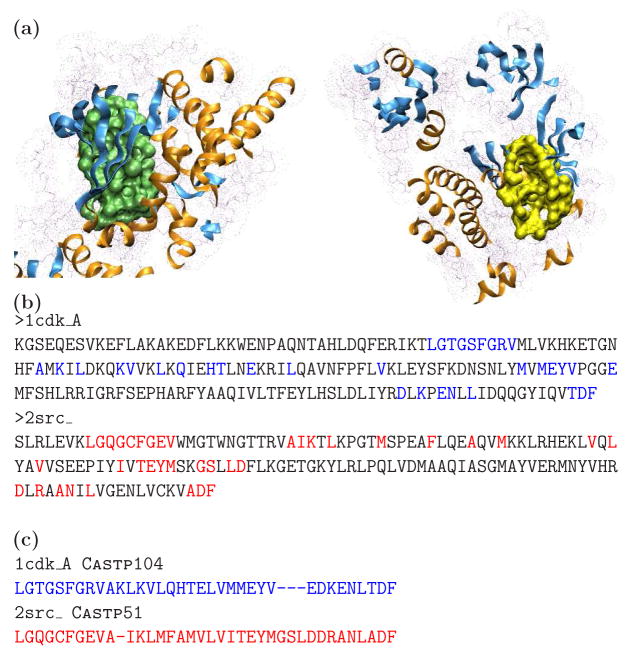
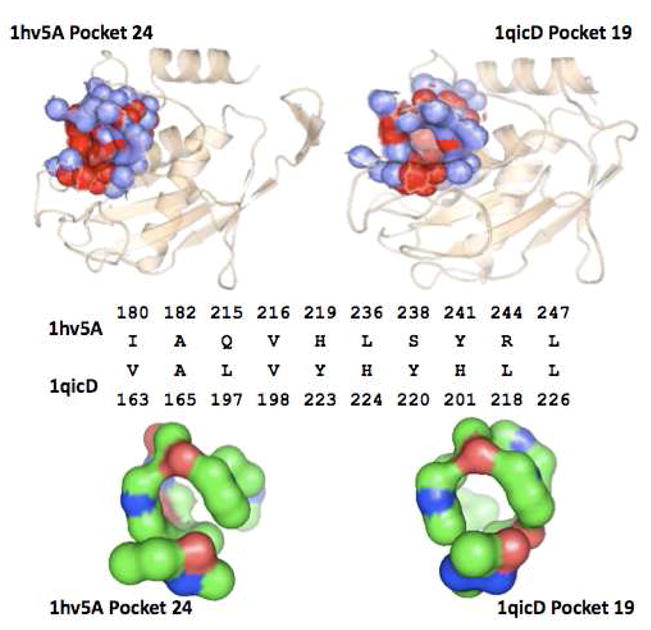
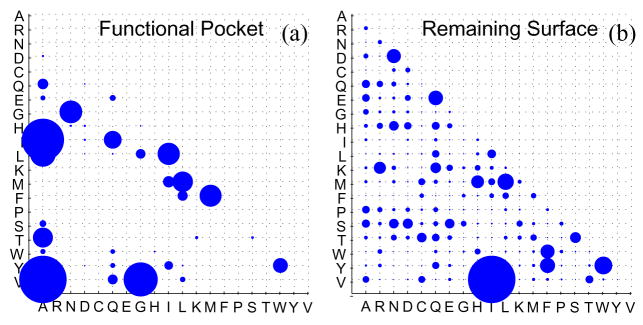
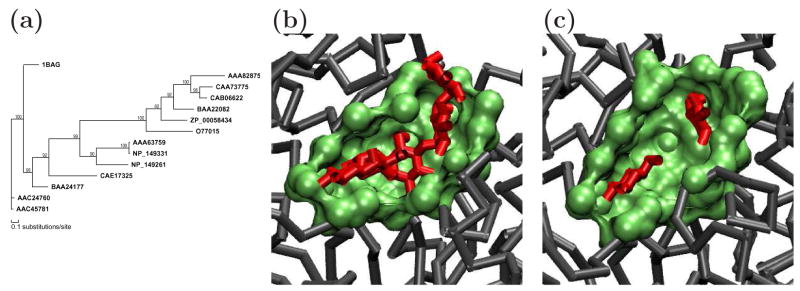
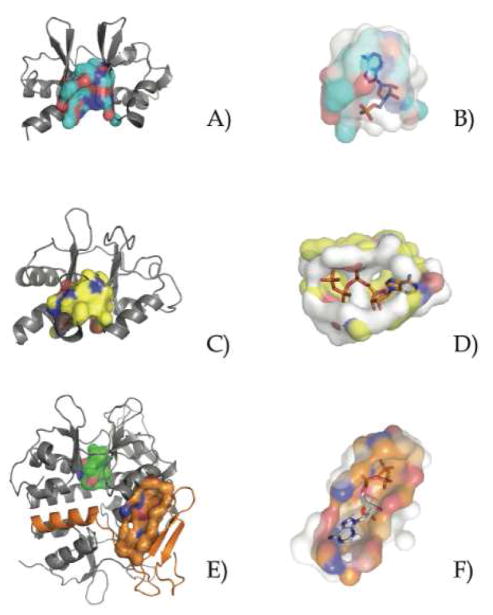
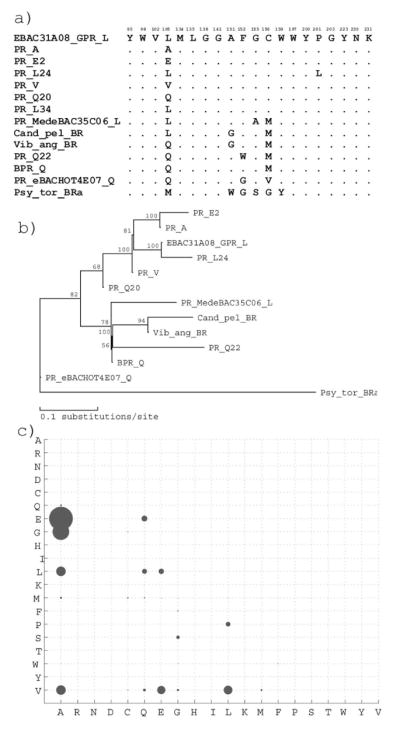
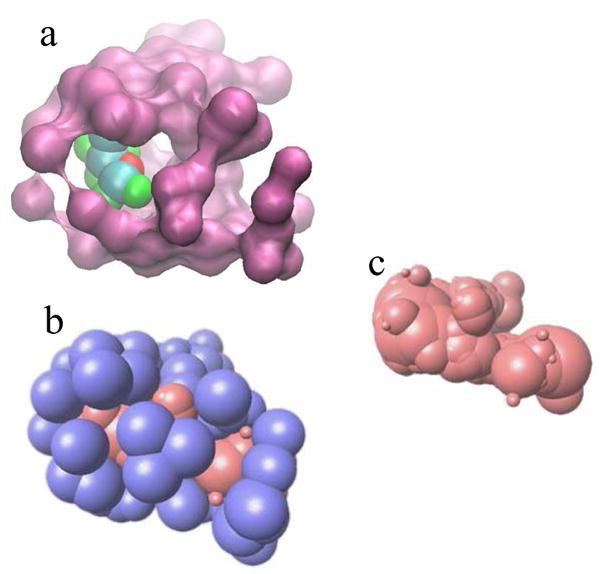
Similar articles
-
pvSOAR: detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins.Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W555-8. doi: 10.1093/nar/gkh390. Nucleic Acids Res. 2004. PMID: 15215448 Free PMC article.
-
Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns.J Mol Biol. 2009 Mar 27;387(2):451-64. doi: 10.1016/j.jmb.2008.12.072. Epub 2009 Jan 6. J Mol Biol. 2009. PMID: 19154742 Free PMC article.
-
Predicting gene ontology functions from protein's regional surface structures.BMC Bioinformatics. 2007 Dec 11;8:475. doi: 10.1186/1471-2105-8-475. BMC Bioinformatics. 2007. PMID: 18070366 Free PMC article.
-
Computational advances in discovering cryptic pockets for drug discovery.Curr Opin Struct Biol. 2025 Feb;90:102975. doi: 10.1016/j.sbi.2024.102975. Epub 2025 Jan 7. Curr Opin Struct Biol. 2025. PMID: 39778412 Review.
-
[Development and validation of programs for ligand-binding-pocket search].Yakugaku Zasshi. 2011;131(10):1429-35. doi: 10.1248/yakushi.131.1429. Yakugaku Zasshi. 2011. PMID: 21963969 Review. Japanese.
Cited by
-
Accuracy of functional surfaces on comparatively modeled protein structures.J Struct Funct Genomics. 2011 Jul;12(2):97-107. doi: 10.1007/s10969-011-9109-z. Epub 2011 May 4. J Struct Funct Genomics. 2011. PMID: 21541664 Free PMC article.
-
Geometry of protein shape and its evolutionary pattern for function prediction and characterization.Annu Int Conf IEEE Eng Med Biol Soc. 2009;2009:2324-7. doi: 10.1109/IEMBS.2009.5335112. Annu Int Conf IEEE Eng Med Biol Soc. 2009. PMID: 19965177 Free PMC article.
-
Computer-Aided Protein Directed Evolution: a Review of Web Servers, Databases and other Computational Tools for Protein Engineering.Comput Struct Biotechnol J. 2012 Oct 22;2:e201209008. doi: 10.5936/csbj.201209008. eCollection 2012. Comput Struct Biotechnol J. 2012. PMID: 24688649 Free PMC article. Review.
References
-
- Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311(5759):347–51. - PubMed
-
- Rost B. Enzyme function less conserved than anticipated. J Mol Biol. 2002;318:595–608. - PubMed
-
- Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol. 2003;333:863–882. - PubMed
-
- Russell RB. Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution. J Mol Biol. 1998;279:1211–1227. - PubMed
-
- Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J Mol Biol. 2003;332:505–526. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
