

Protein misfolding induces hypoxic preconditioning via a subset of the unfolded protein response machinery
- PMID: 20733002
- PMCID: PMC2953055
- DOI: 10.1128/MCB.00922-10
Protein misfolding induces hypoxic preconditioning via a subset of the unfolded protein response machinery
Abstract
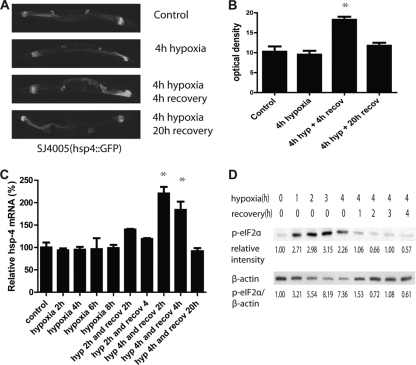
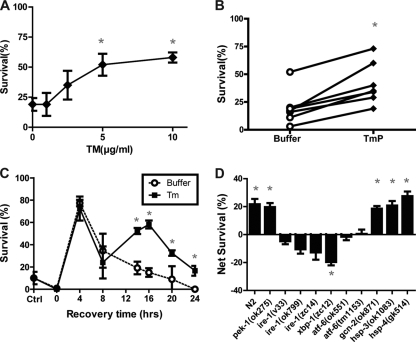
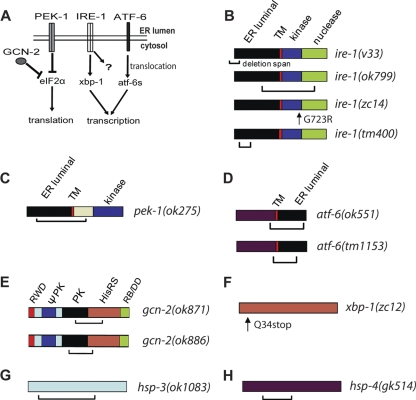
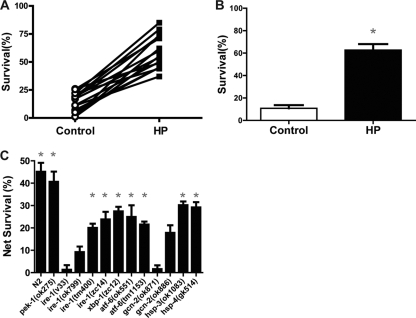
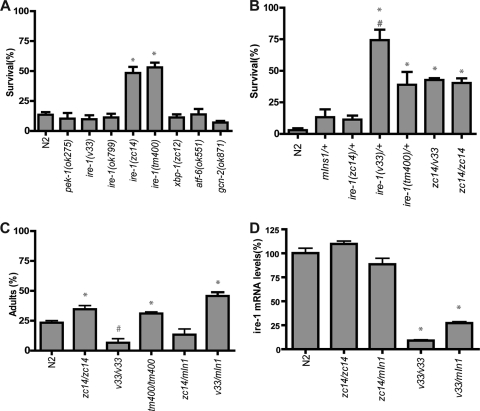
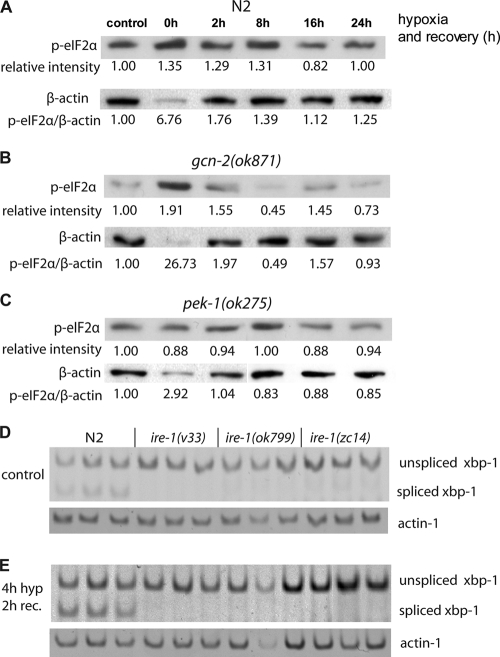
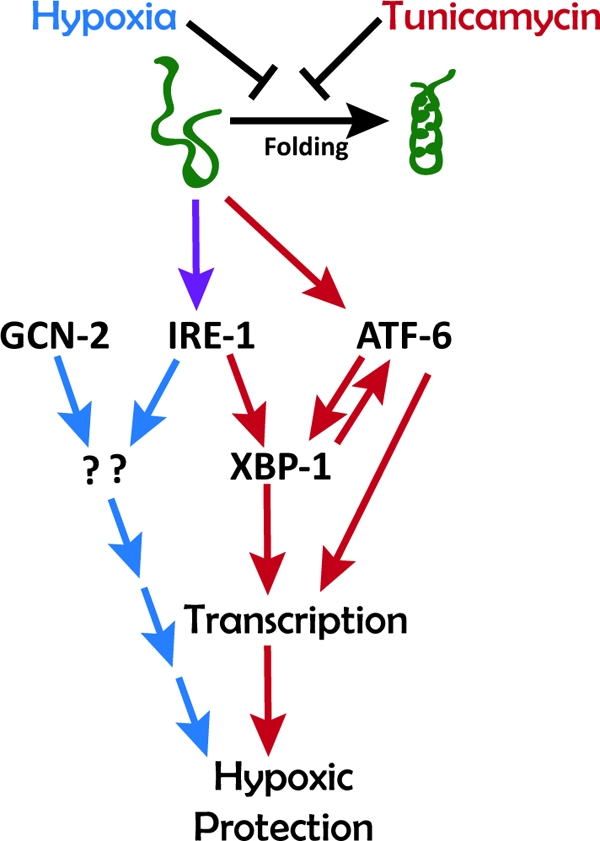
Prolonged cellular hypoxia results in energy failure and ultimately cell death. However, less-severe hypoxia can induce a cytoprotective response termed hypoxic preconditioning (HP). The unfolded protein response pathway (UPR) has been known for some time to respond to hypoxia and regulate hypoxic sensitivity; however, the role of the UPR, if any, in HP essentially has been unexplored. We have shown previously that a sublethal hypoxic exposure of the nematode Caenorhabditis elegans induces a protein chaperone component of the UPR (L. L. Anderson, X. Mao, B. A. Scott, and C. M. Crowder, Science 323:630-633, 2009). Here, we show that HP induces the UPR and that the pharmacological induction of misfolded proteins is itself sufficient to stimulate a delayed protective response to hypoxic injury that requires the UPR pathway proteins IRE-1, XBP-1, and ATF-6. HP also required IRE-1 but not XBP-1 or ATF-6; instead, GCN-2, which is known to suppress translation and induce an adaptive transcriptional response under conditions of UPR activation or amino acid deprivation, was required for HP. The phosphorylation of the translation factor eIF2α, an established mechanism of GCN-2-mediated translational suppression, was not necessary for HP. These data suggest a model where hypoxia-induced misfolded proteins trigger the activation of IRE-1, which along with GCN-2 controls an adaptive response that is essential to HP.
Figures
Similar articles
-
IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA.Nature. 2002 Jan 3;415(6867):92-6. doi: 10.1038/415092a. Nature. 2002. PMID: 11780124
-
The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans.J Cell Sci. 2013 Sep 15;126(Pt 18):4136-46. doi: 10.1242/jcs.123000. Epub 2013 Jul 10. J Cell Sci. 2013. PMID: 23843615 Free PMC article.
-
Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans.PLoS Genet. 2005 Sep;1(3):e37. doi: 10.1371/journal.pgen.0010037. PLoS Genet. 2005. PMID: 16184190 Free PMC article.
-
Hypoxia signaling and resistance in C. elegans.Trends Endocrinol Metab. 2010 Jul;21(7):435-40. doi: 10.1016/j.tem.2010.02.006. Epub 2010 Mar 23. Trends Endocrinol Metab. 2010. PMID: 20335046 Review.
-
Links between the unfolded protein response and the DNA damage response in hypoxia: a systematic review.Biochem Soc Trans. 2021 Jun 30;49(3):1251-1263. doi: 10.1042/BST20200861. Biochem Soc Trans. 2021. PMID: 34003246 Free PMC article.
Cited by
-
Identification of enzymes that have helminth-specific active sites and are required for Rhodoquinone-dependent metabolism as targets for new anthelmintics.PLoS Negl Trop Dis. 2021 Nov 29;15(11):e0009991. doi: 10.1371/journal.pntd.0009991. eCollection 2021 Nov. PLoS Negl Trop Dis. 2021. PMID: 34843467 Free PMC article.
-
Conserved roles of C. elegans and human MANFs in sulfatide binding and cytoprotection.Nat Commun. 2018 Mar 1;9(1):897. doi: 10.1038/s41467-018-03355-0. Nat Commun. 2018. PMID: 29497057 Free PMC article.
-
Nicotinamide mononucleotide adenylyltransferase promotes hypoxic survival by activating the mitochondrial unfolded protein response.Cell Death Dis. 2016 Feb 25;7(2):e2113. doi: 10.1038/cddis.2016.5. Cell Death Dis. 2016. PMID: 26913604 Free PMC article.
-
Ischemic preconditioning: the role of mitochondria and aging.Exp Gerontol. 2012 Jan;47(1):1-7. doi: 10.1016/j.exger.2011.11.001. Epub 2011 Nov 10. Exp Gerontol. 2012. PMID: 22100642 Free PMC article. Review.
-
Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells.PLoS One. 2013 Aug 15;8(8):e73267. doi: 10.1371/journal.pone.0073267. eCollection 2013. PLoS One. 2013. PMID: 24039668 Free PMC article.
References
-
- Bi, M., C. Naczki, M. Koritzinsky, D. Fels, J. Blais, N. Hu, H. Harding, I. Novoa, M. Varia, J. Raleigh, D. Scheuner, R. J. Kaufman, J. Bell, D. Ron, B. G. Wouters, and C. Koumenis. 2005. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 24:3470-3481. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous