

Cystinosis: practical tools for diagnosis and treatment
- PMID: 20734088
- PMCID: PMC3016220
- DOI: 10.1007/s00467-010-1627-6
Cystinosis: practical tools for diagnosis and treatment
Abstract
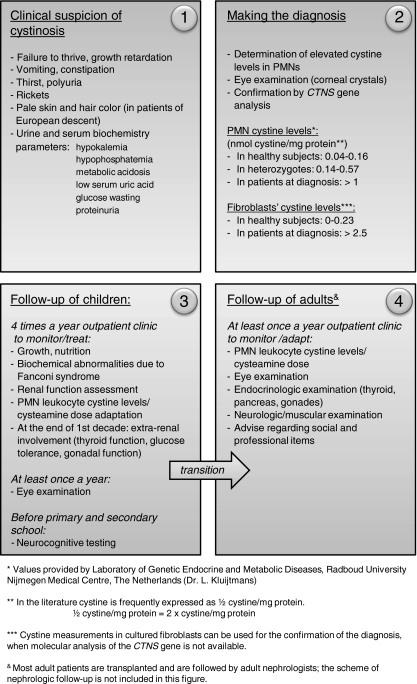
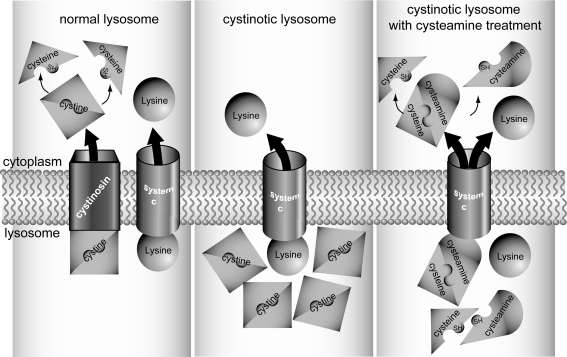
Cystinosis is the major cause of inherited Fanconi syndrome, and should be suspected in young children with failure to thrive and signs of renal proximal tubular damage. The diagnosis can be missed in infants, because not all signs of renal Fanconi syndrome are present during the first months of life. In older patients cystinosis can mimic idiopathic nephrotic syndrome due to focal and segmental glomerulosclerosis. Measuring elevated white blood cell cystine content is the corner stone for the diagnosis. The diagnosis is confirmed by molecular analysis of the cystinosin gene. Corneal cystine crystals are invariably present in all patients with cystinosis after the age of 1 year. Treatment with the cystine depleting drug cysteamine should be initiated as soon as possible and continued lifelong to prolong renal function survival and protect extra-renal organs. This educational feature provides practical tools for the diagnosis and treatment of cystinosis.
Figures
References
-
- Fanconi G, Bickel H. Die chronische Aminoacidurie (Aminosäurediabetes oder nephrotisch-glukosurischer Zwergwuchs) bei der Glykogenose und der Cystinkrankheit. Helv Paediatr Acta. 1949;4:359–396. - PubMed
-
- Brodehl J, Hagge W, Gellissen K. Changes in kidney function in cystinosis. I. Inulin, PAH and electrolyte clearance in various stages of the disease. Ann Paediatr. 1965;205:131–154. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
