

Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury
- PMID: 20738443
- PMCID: PMC3823156
- DOI: 10.1111/j.1582-4934.2010.01164.x
Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury
Abstract
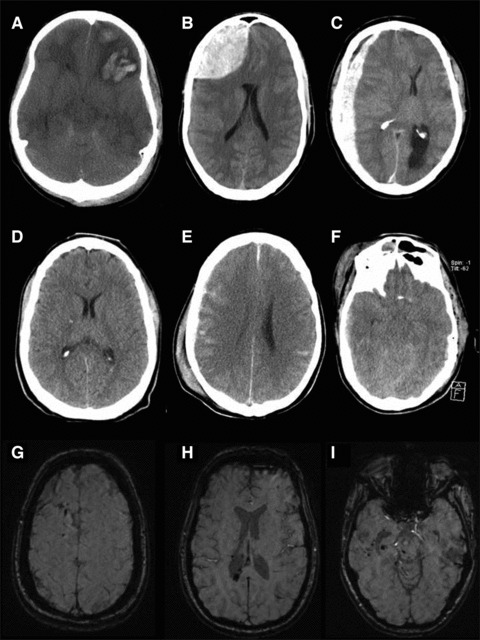
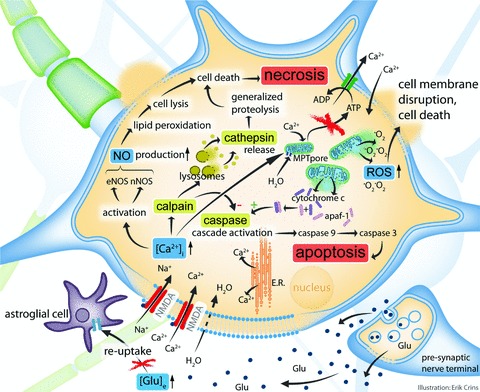
Traumatic brain injury (TBI) is a frequent and clinically highly heterogeneous neurological disorder with large socioeconomic consequences. TBI severity classification, based on the hospital admission Glasgow Coma Scale (GCS) score, ranges from mild (GCS 13-15) and moderate (GCS 9-12) to severe (GCS ≤ 8). The GCS reflects the risk of dying from TBI, which is low after mild (∼1%), intermediate after moderate (up to 15%) and high (up to 40%) after severe TBI. Intracranial damage can be focal, such as epidural and subdural haematomas and parenchymal contusions, or diffuse, for example traumatic axonal injury and diffuse cerebral oedema, although this distinction is somewhat arbitrary. Study of the cellular and molecular post-traumatic processes is essential for the understanding of TBI pathophysiology but even more to find therapeutic targets for the development of neuroprotective drugs to be eventually used in human beings. To date, studies in vitro and in vivo, mainly in animals but also in human beings, are unravelling the pathological TBI mechanisms at high pace. Nevertheless, TBI pathophysiology is all but completely elucidated. Neuroprotective treatment studies in human beings have been disappointing thus far and have not resulted in commonly accepted drugs. This review presents an overview on the clinical aspects and the pathophysiology of focal and diffuse TBI, and it highlights several acknowledged important events that occur on molecular and cellular level after TBI.
© 2010 The Authors Journal compilation © 2010 Foundation for Cellular and Molecular Medicine/Blackwell Publishing Ltd.
Figures

References
-
- Tagliaferri F, Compagnone C, Korsic M, et al. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 2006;148:255–68. - PubMed
-
- Rutland-Brown W, Langlois JA, Thomas KE, et al. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil. 2006;21:544–8. - PubMed
-
- Murray GD, Teasdale GM, Braakman R, et al. The European Brain Injury Consortium survey of head injuries. Acta Neurochir. 1999;141:223–36. - PubMed
-
- Cremer OL, Moons KG, Van Dijk GW, et al. Prognosis following severe head injury: development and validation of a model for prediction of death, disability, and functional recovery. J Trauma. 2006;61:1484–91. - PubMed
-
- Myburgh JA, Cooper DJ, Finfer SR, et al. Epidemiology and 12-month outcomes from traumatic brain injury in australia and new zealand. J Trauma. 2008;64:854–62. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
