

Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex
- PMID: 20739563
- PMCID: PMC3842472
- DOI: 10.1523/JNEUROSCI.2127-10.2010
Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex
Abstract
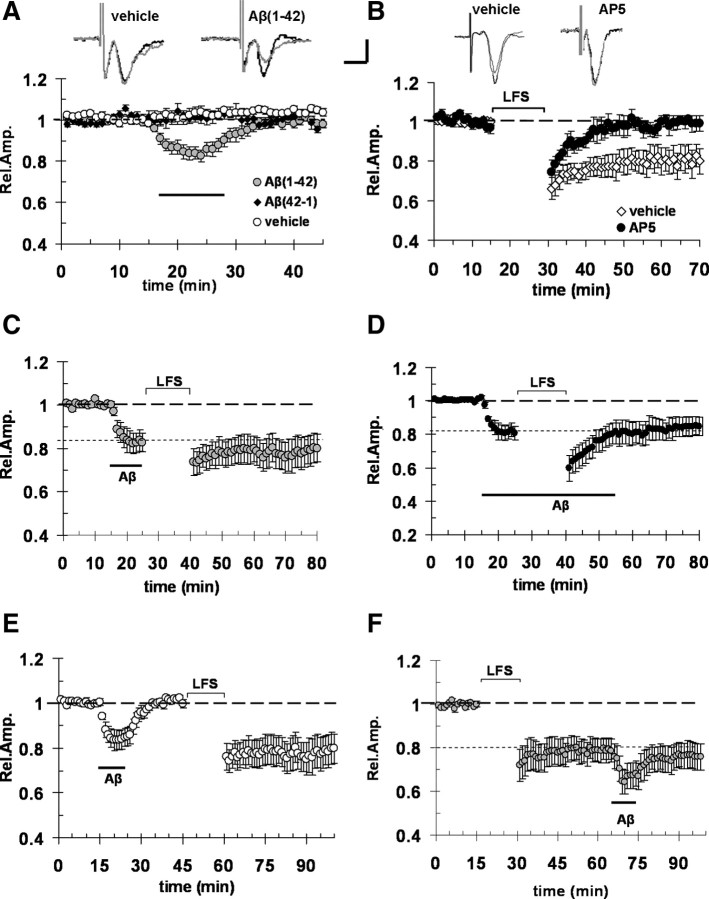
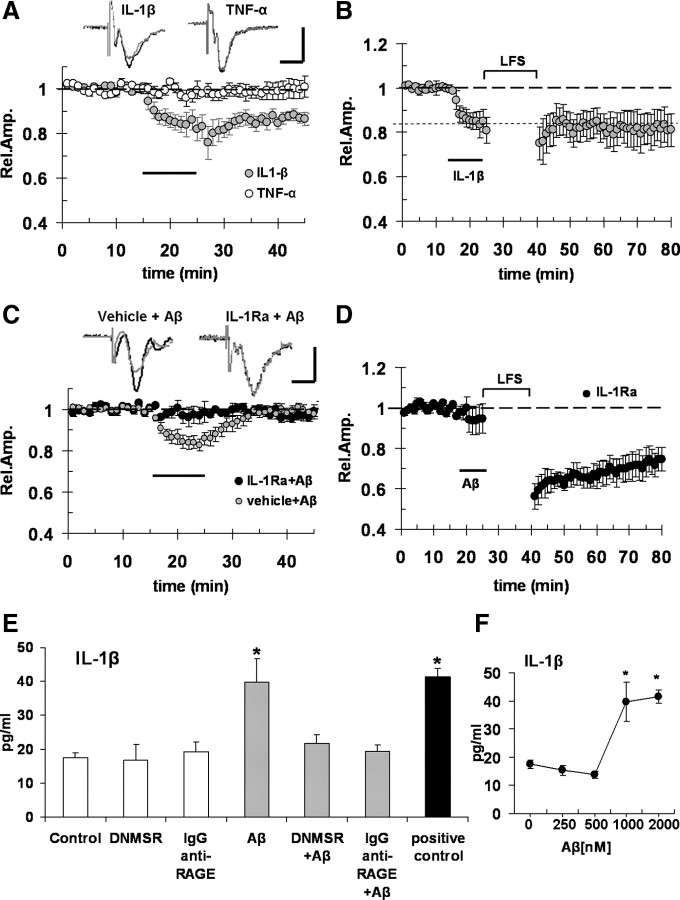
Overproduction of beta-amyloid (Abeta) is a pathologic feature of Alzheimer's disease, leading to cognitive impairment. Here, we investigated the impact of cell-specific receptor for advanced glycation end products (RAGE) on Abeta-induced entorhinal cortex (EC) synaptic dysfunction. We found both a transient depression of basal synaptic transmission and inhibition of long-term depression (LTD) after the application of Abeta in EC slices. Synaptic depression and LTD impairment induced by Abeta were rescued by functional suppression of RAGE. Remarkably, the rescue was only observed in slices from mice expressing a defective form of RAGE targeted to microglia, but not in slices from mice expressing defective RAGE targeted to neurons. Moreover, we found that the inflammatory cytokine IL-1beta (interleukin-1beta) and stress-activated kinases [p38 MAPK (p38 mitogen-activated protein kinase) and JNK (c-Jun N-terminal kinase)] were significantly altered and involved in RAGE signaling pathways depending on RAGE expression in neuron or microglia. These findings suggest a prominent role of microglial RAGE signaling in Abeta-induced EC synaptic dysfunction.
Figures




References
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. - PMC - PubMed
-
- Anderson WW, Collingridge GL. The LTP program: a data acquisition program for on-line analysis of long-term potentiation and other synaptic events. J Neurosci Methods. 2001;108:71–83. - PubMed
-
- Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23:4096–4105. - PMC - PubMed
-
- Bianchi R, Giambanco I, Donato R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging. 2010;31:665–677. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous