

The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes
- PMID: 20739713
- PMCID: PMC2975287
- DOI: 10.1534/genetics.110.118661
The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes
Abstract
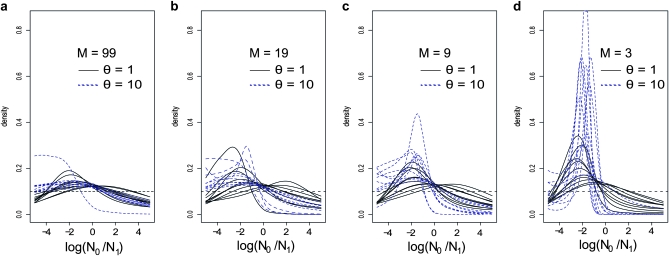
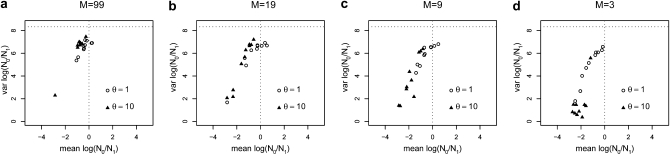
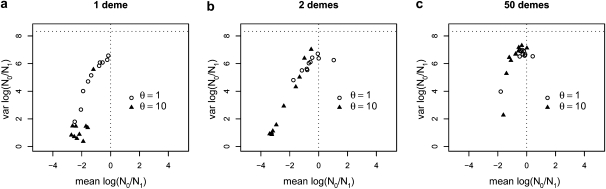
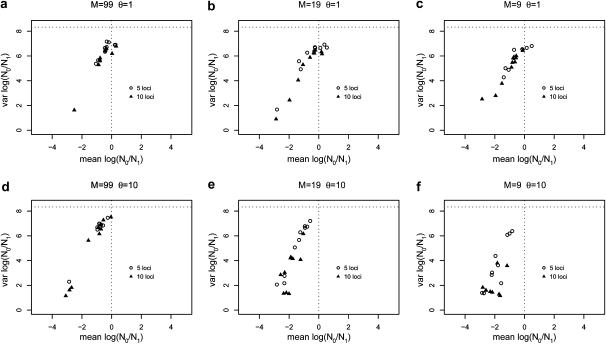
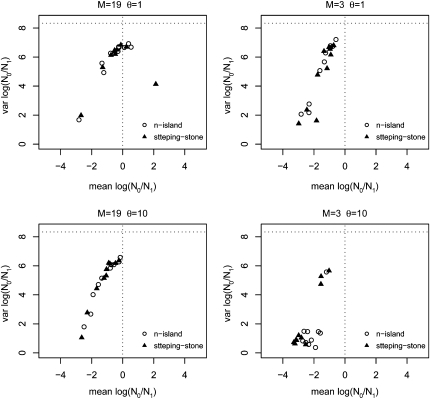
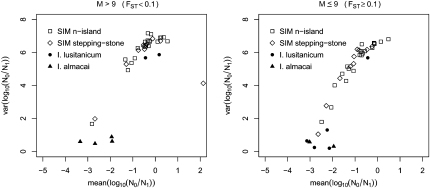
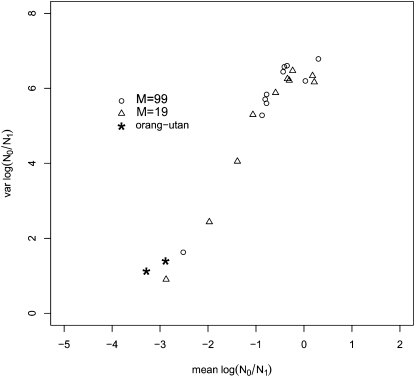
The idea that molecular data should contain information on the recent evolutionary history of populations is rather old. However, much of the work carried out today owes to the work of the statisticians and theoreticians who demonstrated that it was possible to detect departures from equilibrium conditions (e.g., panmictic population/mutation-drift equilibrium) and interpret them in terms of deviations from neutrality or stationarity. During the last 20 years the detection of population size changes has usually been carried out under the assumption that samples were obtained from populations that can be approximated by a Wright-Fisher model (i.e., assuming panmixia, demographic stationarity, etc.). However, natural populations are usually part of spatial networks and are interconnected through gene flow. Here we simulated genetic data at mutation and migration-drift equilibrium under an n-island and a stepping-stone model. The simulated populations were thus stationary and not subject to any population size change. We varied the level of gene flow between populations and the scaled mutation rate. We also used several sampling schemes. We then analyzed the simulated samples using the Bayesian method implemented in MSVAR, the Markov Chain Monte Carlo simulation program, to detect and quantify putative population size changes using microsatellite data. Our results show that all three factors (genetic differentiation/gene flow, genetic diversity, and the sampling scheme) play a role in generating false bottleneck signals. We also suggest an ad hoc method to counter this effect. The confounding effect of population structure and of the sampling scheme has practical implications for many conservation studies. Indeed, if population structure is creating "spurious" bottleneck signals, the interpretation of bottleneck signals from genetic data might be less straightforward than it would seem, and several studies may have overestimated or incorrectly detected bottlenecks in endangered species.
Figures
References
-
- Alves, M. J., and M. M. Coelho, 1994. Genetic variation and population subdivision of the endangered iberian cyprinid chondrostoma lusitanicum. J. Fish Biol. 44 627–636.
-
- Beaumont, M., and R. Nichols, 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B Biol. Sci. 263 1619–1626.
-
- Beaumont, M. A., 2003. b Conservation genetics, pp. 751–792 in Handbook of Statistical Genetics, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, New York.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
