

cAMP-activated Ca2+ signaling is required for CFTR-mediated serous cell fluid secretion in porcine and human airways
- PMID: 20739756
- PMCID: PMC2929731
- DOI: 10.1172/JCI42992
cAMP-activated Ca2+ signaling is required for CFTR-mediated serous cell fluid secretion in porcine and human airways
Abstract
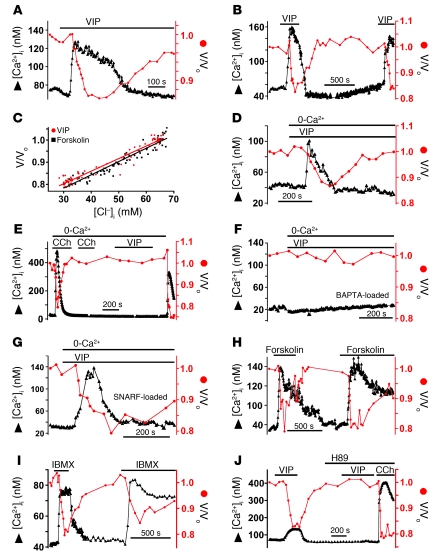
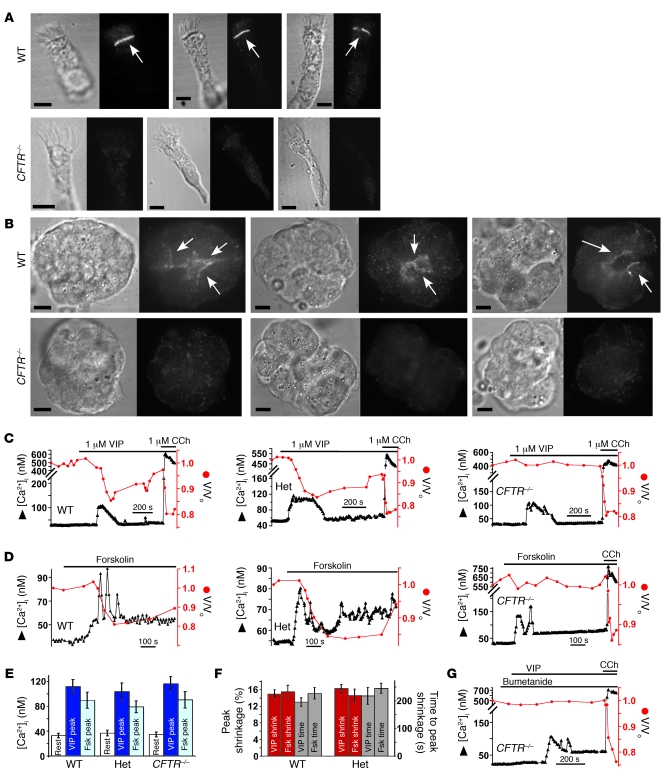
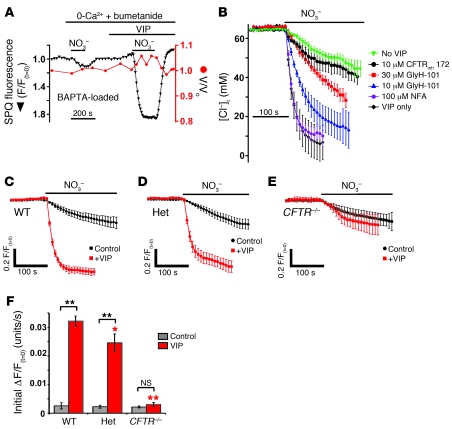
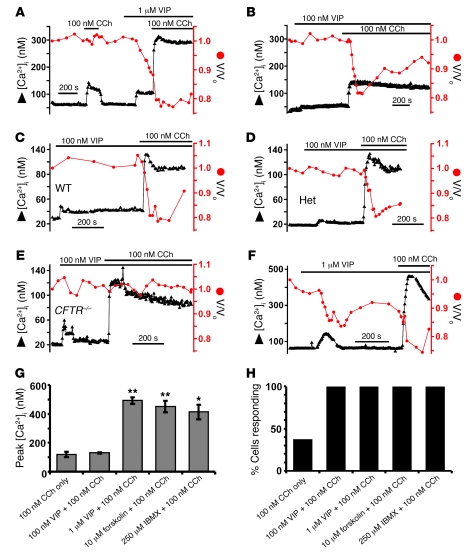
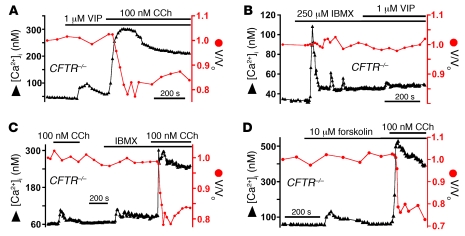
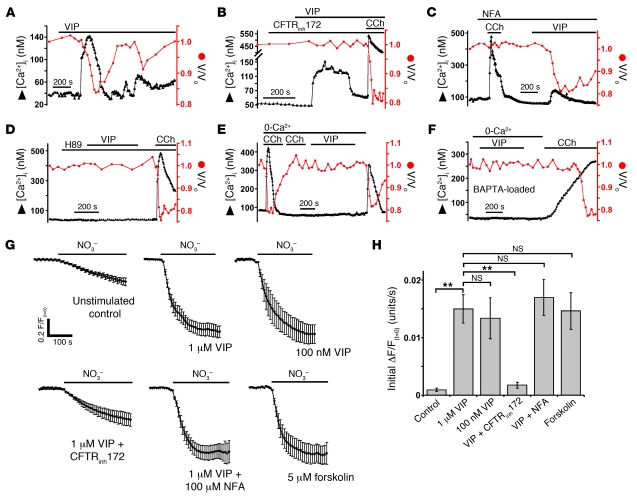
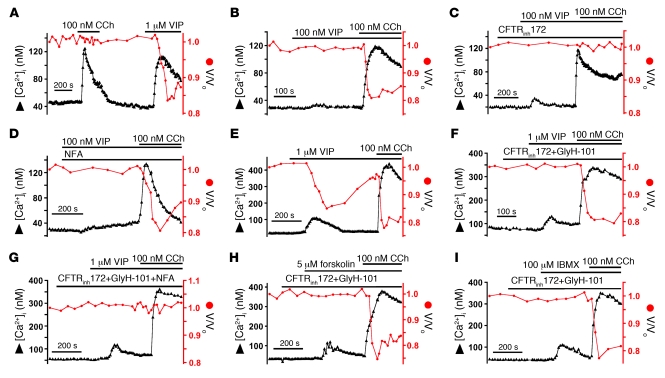
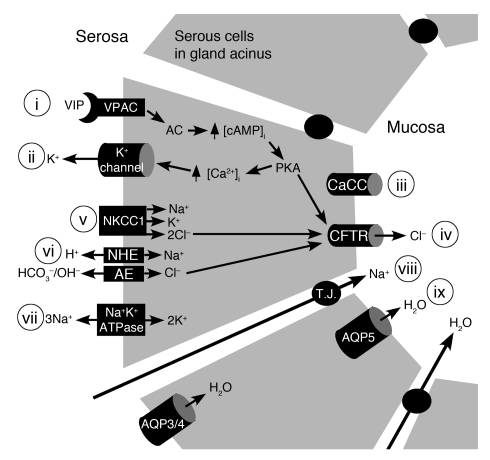
Cystic fibrosis (CF), which is caused by mutations in CFTR, affects many tissues, including the lung. Submucosal gland serous acinar cells are primary sites of fluid secretion and CFTR expression in the lung. Absence of CFTR in these cells may contribute to CF lung pathogenesis by disrupting fluid secretion. Here, we have isolated primary serous acinar cells from wild-type and CFTR-/- pigs and humans without CF to investigate the cellular mechanisms and regulation of fluid secretion by optical imaging. Porcine and human serous cells secrete fluid in response to vasoactive intestinal polypeptide (VIP) and other agents that raise intracellular cAMP levels; here, we have demonstrated that this requires CFTR and a cAMP-dependent rise in intracellular Ca2+ concentration ([Ca2+]i). Importantly, cAMP induced the release of Ca2+ from InsP3-sensitive Ca2+ stores also responsive to cAMP-independent agonists such as cholinergic, histaminergic, and purinergic agonists that stimulate CFTR-independent fluid secretion. This provides two types of synergism that strongly potentiated cAMP-mediated fluid secretion but differed in their CFTR dependencies. First, CFTR-dependent secretion was strongly potentiated by low VIP and carbachol concentrations that individually were unable to stimulate secretion. Second, higher VIP concentrations more strongly potentiated the [Ca2+]i responses, enabling ineffectual levels of cholinergic stimulation to strongly activate CFTR-independent fluid secretion. These results identify important molecular mechanisms of cAMP-dependent secretion, including a requirement for Ca2+ signaling, and suggest new therapeutic approaches to correct defective submucosal gland secretion in CF.
Figures
Comment in
-
Transgenic animals may help resolve a sticky situation in cystic fibrosis.J Clin Invest. 2010 Sep;120(9):3093-6. doi: 10.1172/JCI44235. Epub 2010 Aug 25. J Clin Invest. 2010. PMID: 20739746 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
