

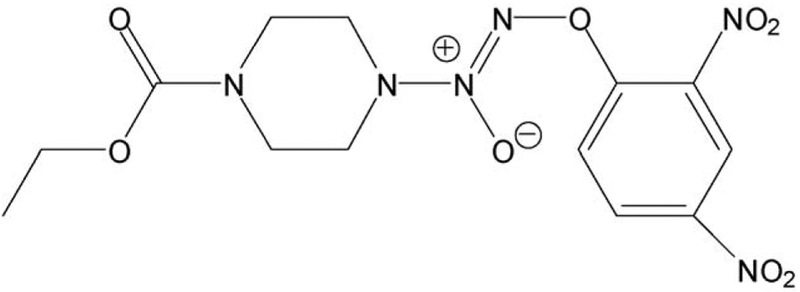
JS-K, a nitric oxide-releasing prodrug, modulates ß-catenin/TCF signaling in leukemic Jurkat cells: evidence of an S-nitrosylated mechanism
- PMID: 20797387
- PMCID: PMC6959133
- DOI: 10.1016/j.bcp.2010.08.011
JS-K, a nitric oxide-releasing prodrug, modulates ß-catenin/TCF signaling in leukemic Jurkat cells: evidence of an S-nitrosylated mechanism
Abstract
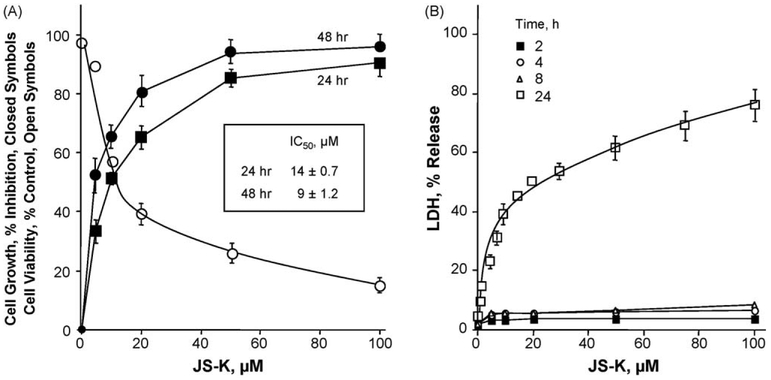
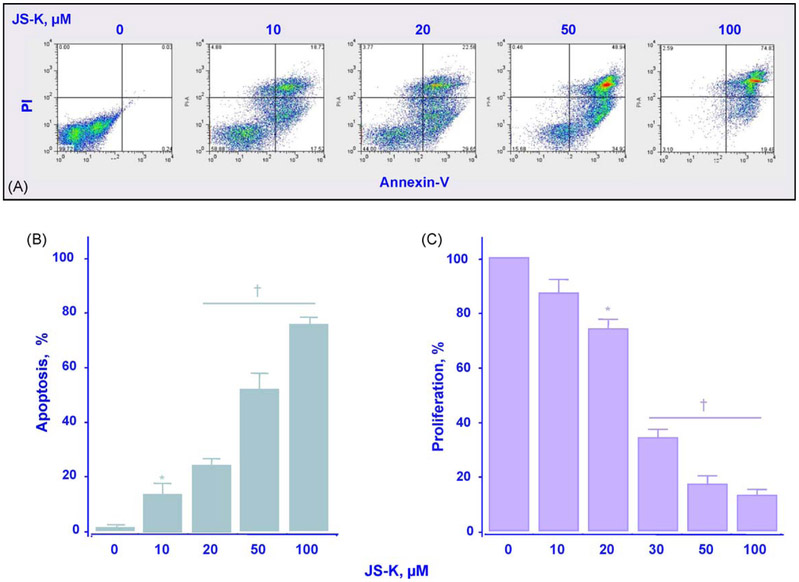
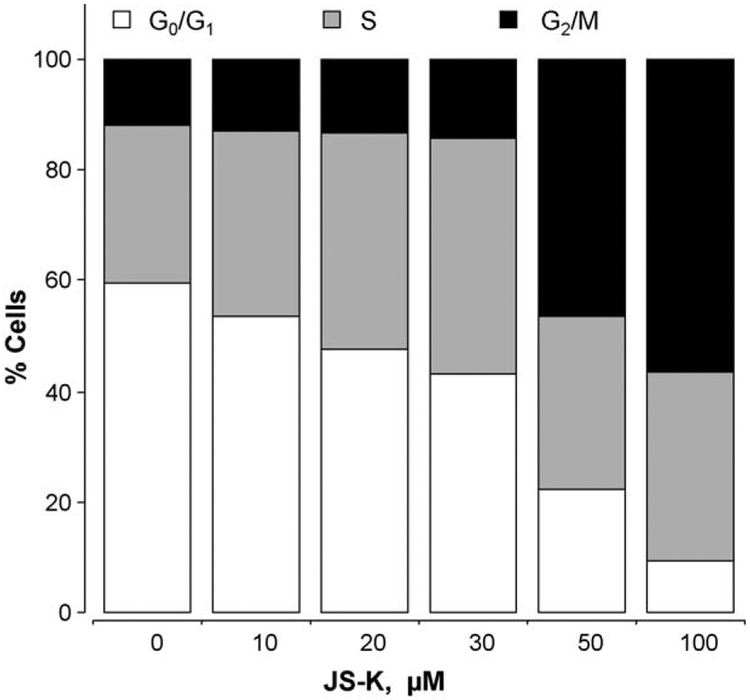
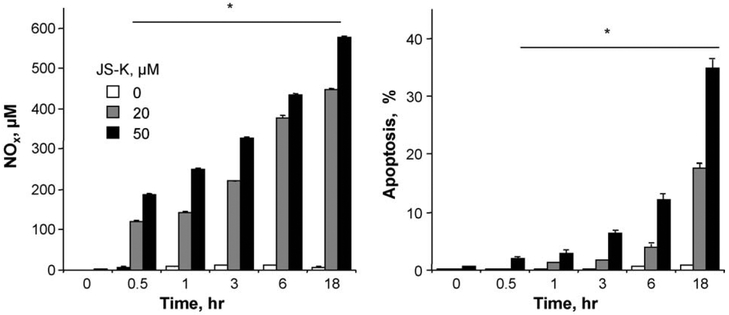
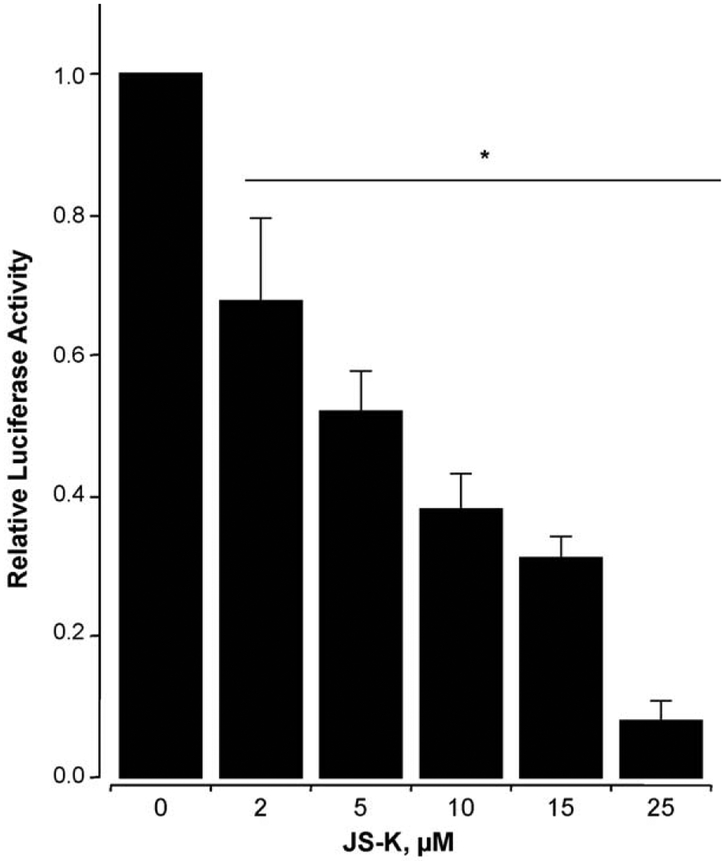
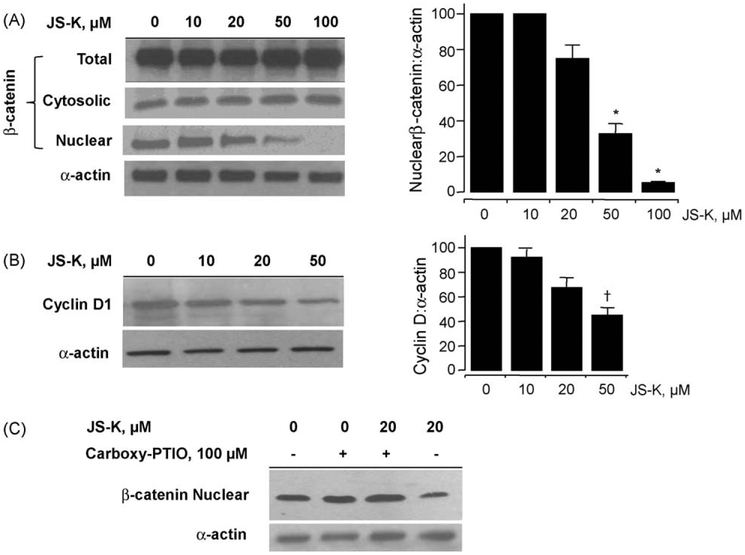
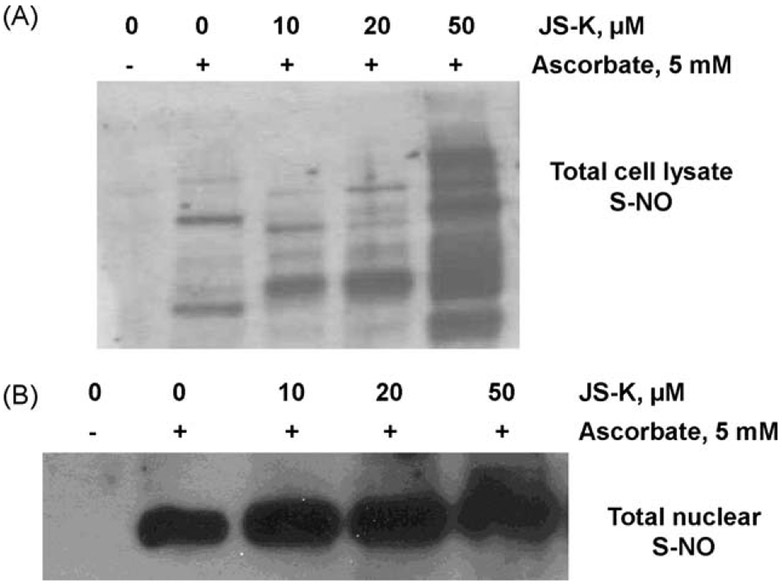
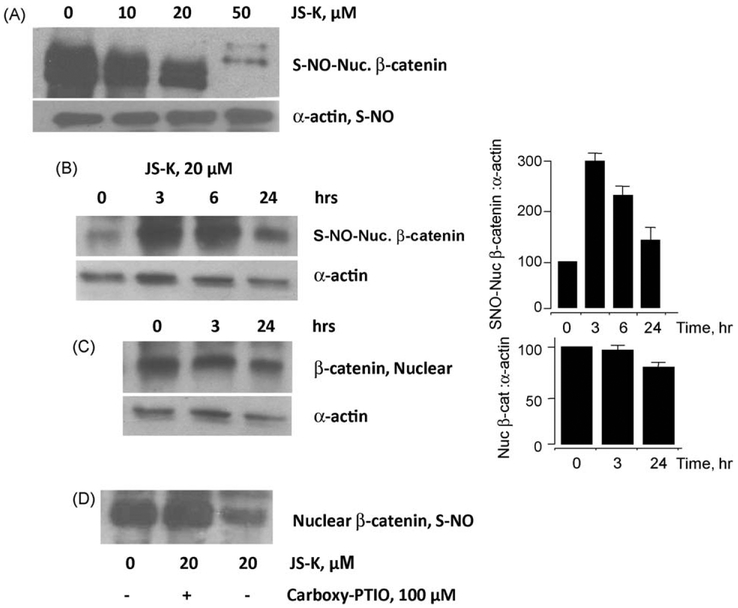
β-Catenin is a central player of the Wnt signaling pathway that regulates cell-cell adhesion and may promote leukemia cell proliferation. We examined whether JS-K, an NO-donating prodrug, modulates the Wnt/β-catenin/TCF-4 signaling pathway in Jurkat T-Acute Lymphoblastic Leukemia cells. JS-K inhibited Jurkat T cell growth in a concentration and time-dependent manner. The IC(50)s for cell growth inhibition were 14±0.7 and 9±1.2μM at 24 and 48h, respectively. Treatment of the cells with JS-K for 24h, caused a dose-dependent increase in apoptosis from 16±3.3% at 10μM to 74.8±2% at 100μM and a decrease in proliferation. This growth inhibition was also due, in part, to alterations in the different phases of the cell cycle. JS-K exhibited a dose-dependent cytotoxicity as measured by LDH release at 24h. However, between 2 and 8h, LDH release was less than 20% for any indicated JS-K concentration. The β-catenin/TCF-4 transcriptional inhibitory activity was reduced by 32±8, 63±5, and 93±2% at 2, 10, and 25μM JS-K, respectively, based on luciferase reporter assays. JS-K reduced nuclear β-catenin and cyclin D1 protein levels, but cytosolic β-catenin expression did not change. Based on a time-course assay of S-nitrosylation of proteins by a biotin switch assay, S-nitrsolyation of nuclear β-catenin was determined to precede its degradation. A comparison of the S-nitrosylated nuclear β-catenin to the total nuclear β-catenin showed that β-catenin protein levels were degraded at 24h, while S-nitrosylation of β-catenin occurred earlier at 0-6h. The NO scavenger PTIO abrogated the JS-K mediated degradation of β-catenin demonstrating the need for NO.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
JS-K, a glutathione/glutathione S-transferase-activated nitric oxide releasing prodrug inhibits androgen receptor and WNT-signaling in prostate cancer cells.BMC Cancer. 2012 Mar 30;12:130. doi: 10.1186/1471-2407-12-130. BMC Cancer. 2012. PMID: 22462810 Free PMC article.
-
Protein phosphatase 2A activation mechanism contributes to JS-K induced caspase-dependent apoptosis in human hepatocellular carcinoma cells.J Exp Clin Cancer Res. 2018 Jul 9;37(1):142. doi: 10.1186/s13046-018-0823-2. J Exp Clin Cancer Res. 2018. PMID: 29986744 Free PMC article.
-
The Nitric Oxide Prodrug JS-K Induces Ca(2+)-Mediated Apoptosis in Human Hepatocellular Carcinoma HepG2 Cells.J Biochem Mol Toxicol. 2016 Apr;30(4):192-9. doi: 10.1002/jbt.21778. Epub 2015 Nov 30. J Biochem Mol Toxicol. 2016. PMID: 26616367
-
Role of β-catenin/TCF-4 signaling in HIV replication and pathogenesis: insights to informing novel anti-HIV molecular therapeutics.J Neuroimmune Pharmacol. 2011 Jun;6(2):247-59. doi: 10.1007/s11481-011-9266-7. Epub 2011 Mar 8. J Neuroimmune Pharmacol. 2011. PMID: 21384147 Free PMC article. Review.
-
The role of Wnt/beta-catenin signaling in renal carcinogenesis: lessons from cadmium toxicity studies.Curr Mol Med. 2010 Jun;10(4):387-404. doi: 10.2174/156652410791316986. Curr Mol Med. 2010. PMID: 20455852 Review.
Cited by
-
The contribution of N₂O₃ to the cytotoxicity of the nitric oxide donor DETA/NO: an emerging role for S-nitrosylation.Biosci Rep. 2013 Mar 28;33(2):e00031. doi: 10.1042/BSR20120120. Biosci Rep. 2013. PMID: 23402389 Free PMC article.
-
14-aminotetradecanoic acid exhibits antioxidant activity and ameliorates xenobiotics-induced cytotoxicity.Mol Cell Biochem. 2012 May;364(1-2):1-9. doi: 10.1007/s11010-011-1196-4. Epub 2011 Dec 24. Mol Cell Biochem. 2012. PMID: 22198290
-
Novel strategies to target the ubiquitin proteasome system in multiple myeloma.Oncotarget. 2016 Feb 9;7(6):6521-37. doi: 10.18632/oncotarget.6658. Oncotarget. 2016. PMID: 26695547 Free PMC article. Review.
-
Endothelial NO Synthase-Dependent S-Nitrosylation of β-Catenin Prevents Its Association with TCF4 and Inhibits Proliferation of Endothelial Cells Stimulated by Wnt3a.Mol Cell Biol. 2017 May 31;37(12):e00089-17. doi: 10.1128/MCB.00089-17. Print 2017 Jun 15. Mol Cell Biol. 2017. PMID: 28320874 Free PMC article.
-
Nitric Oxide-Releasing Hybrid Drugs Target Cellular Processes Through S-Nitrosylation.For Immunopathol Dis Therap. 2012;3(2):97-108. doi: 10.1615/ForumImmunDisTher.2012006099. For Immunopathol Dis Therap. 2012. PMID: 23730526 Free PMC article.
References
-
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009;59:225–49. - PubMed
-
- Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical significance of cytogenetic abnormalities in adult acute lymphoblastic leukemia. Blood 1998;91:3995–4019. - PubMed
-
- Stefano F, Distrutti E. Cyclo-oxygenase (COX) inhibiting nitric oxide donating (CINODs) drugs: a review of their current status. Curr Top Med Chem 2007;7:277–82. - PubMed
-
- Li CQ, Wogan GN. Nitric oxide as a modulator of apoptosis. Cancer Lett 2005;226:1–15. - PubMed
-
- Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun 2001;282:1075–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
