

Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome
- PMID: 20798252
- PMCID: PMC3001773
- DOI: 10.2215/CJN.01190210
Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome
Erratum in
- Clin J Am Soc Nephrol. 2012 Aug;7(8):1372-4
Abstract
Background and objectives: Mutations in podocyte genes are associated with steroid-resistant nephrotic syndrome (SRNS), mostly affecting younger age groups. To date, it is unclear whether these patients benefit from intensified immunosuppression with cyclosporine A (CsA). The aim of this study was to evaluate the influence of podocyte gene defects in congenital nephrotic syndrome (CNS) and pediatric SRNS on the efficacy of CsA therapy and preservation of renal function.
Design, settings, participants, & measurements: Genotyping was performed in 91 CNS/SRNS patients, irrespective of age at manifestation or response to CsA.
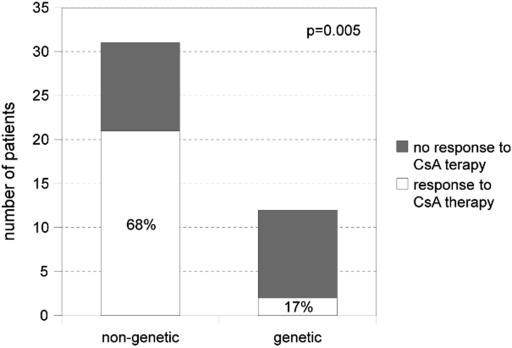
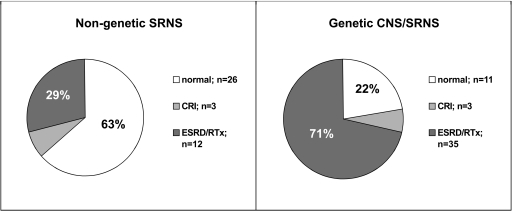
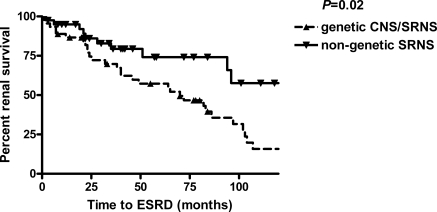
Results: Mutations were identified in 52% of families (11 NPHS1, 17 NPHS2, 11 WT1, 1 LAMB2, 3 TRPC6). Sixty-eight percent of patients with nongenetic SRNS responded to CsA, most of them achieved complete remission. In contrast, none of the patients with genetic CNS/SRNS experienced a complete remission and only two (17%) achieved a partial response, both affected by a WT1 mutation. Preservation of renal function was significantly better in children with nongenetic disease after a mean follow-up time of 8.6 years (ESRD in 29% versus 71%).
Conclusions: The mutation detection rate in our population was high (52%). Most patients with genetic CNS/SRNS did not benefit from CsA with significantly lower response rates compared with nongenetic patients and showed rapid progression to end-stage renal failure. These data strongly support the idea not to expose CNS/SRNS patients with inherited defects related to podocyte function to intensified immunosuppression with CsA.
Figures
References
-
- Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K: Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998 - PubMed
-
- Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler M-C, Niaudet P, Antignac C: NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000 - PubMed
-
- Jeanpierre C, Denamur E, Henry I, Cabanis MO, Luce S, Cécille A, Elion J, Peuchmaur M, Loirat C, Niaudet P, Gubler MC, Junien C: Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am J Hum Genet 62: 824–833, 1998 - PMC - PubMed
-
- Zenker M, Aigner T, Wendler O, Tralau T, Müntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wühl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dötsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A: Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 13: 2625–2632, 2004 - PubMed
-
- Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR: TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37: 739–744, 2005 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
