

Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain
- PMID: 20799731
- PMCID: PMC2943563
- DOI: 10.1021/bi101052d
Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain
Abstract
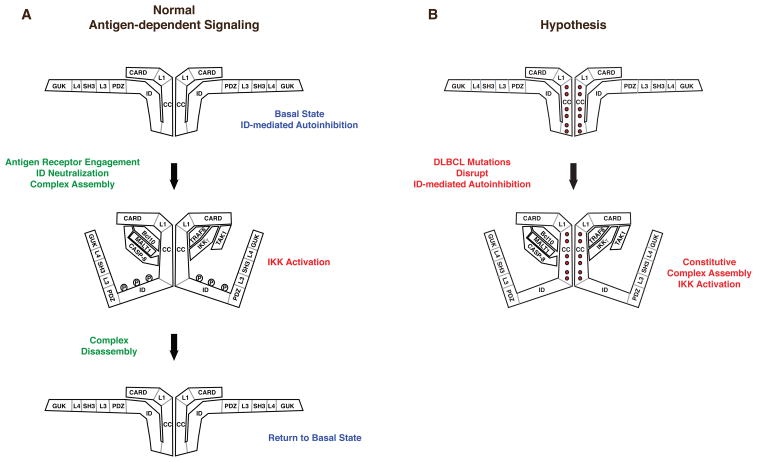
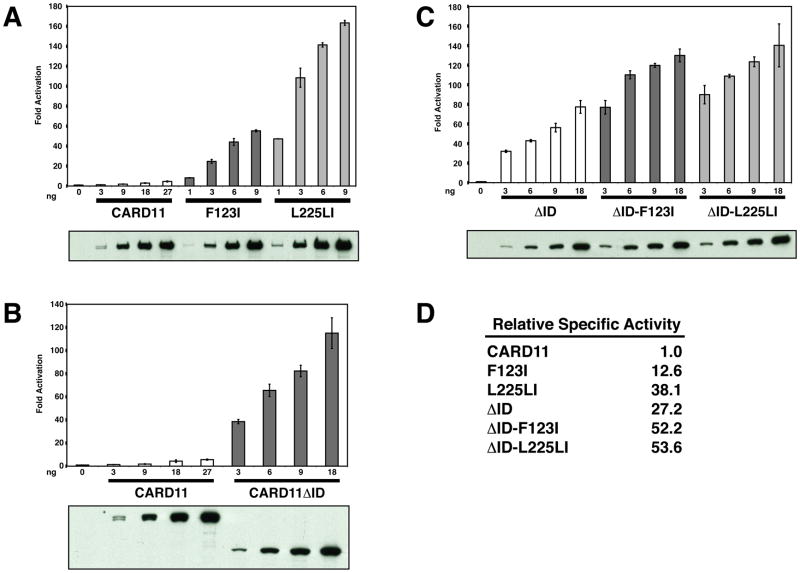
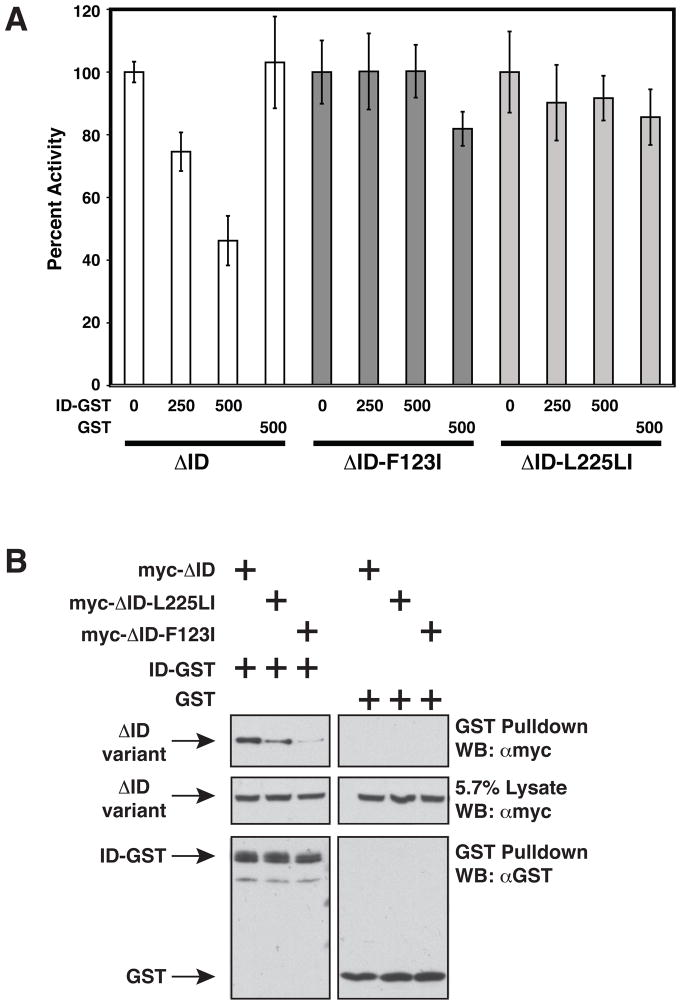
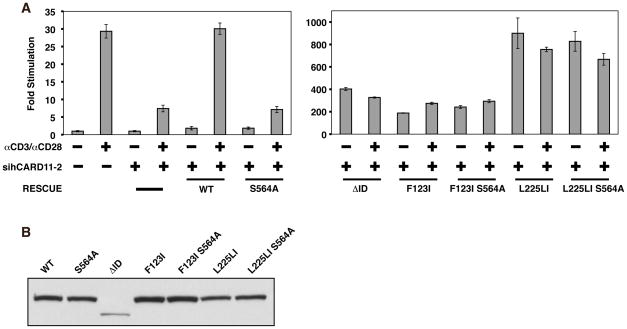
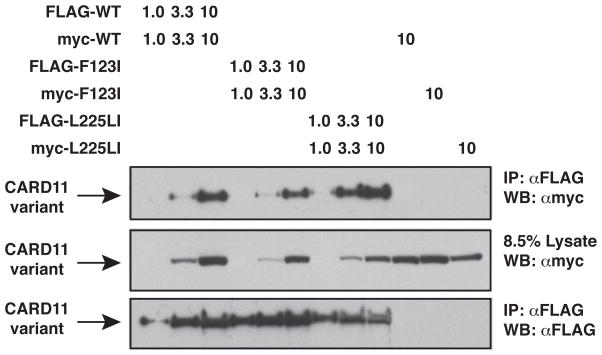
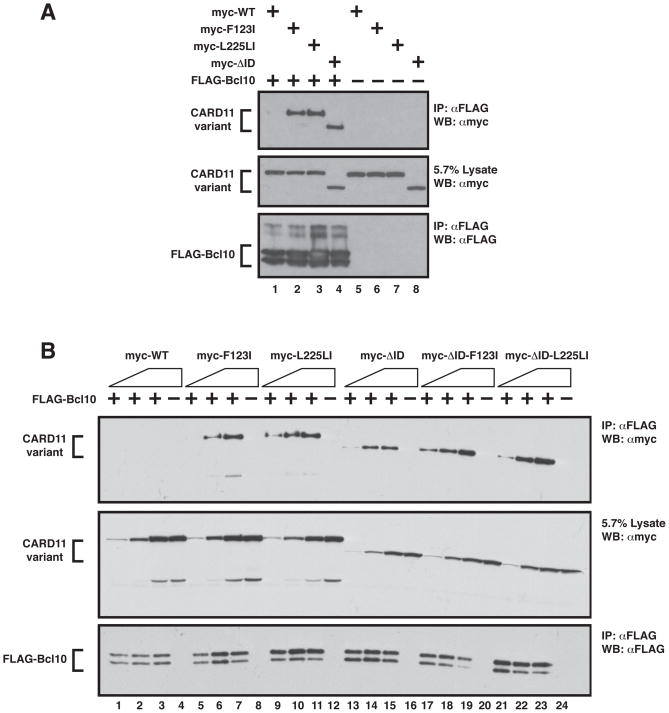
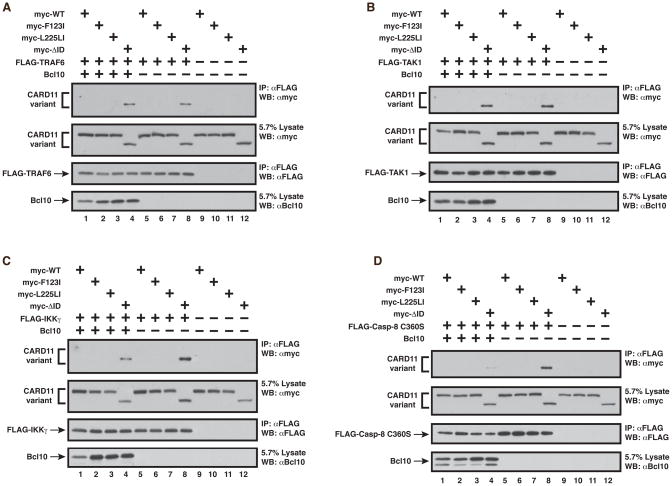
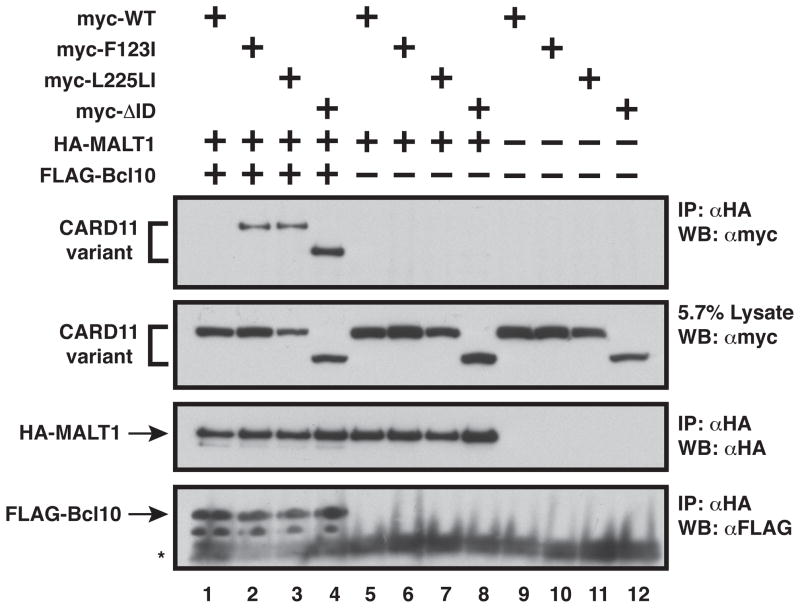
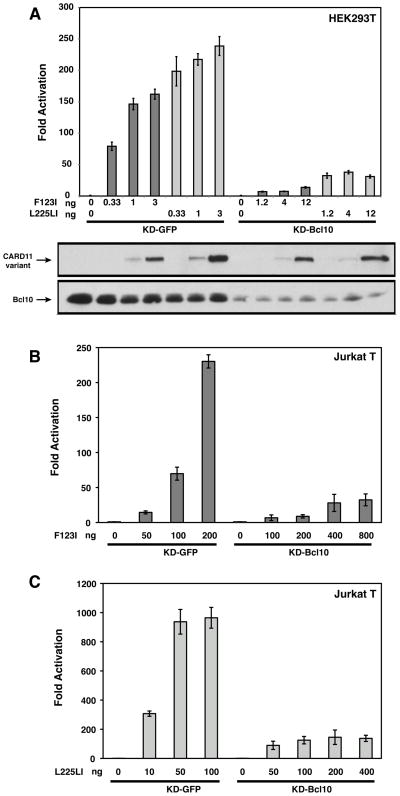
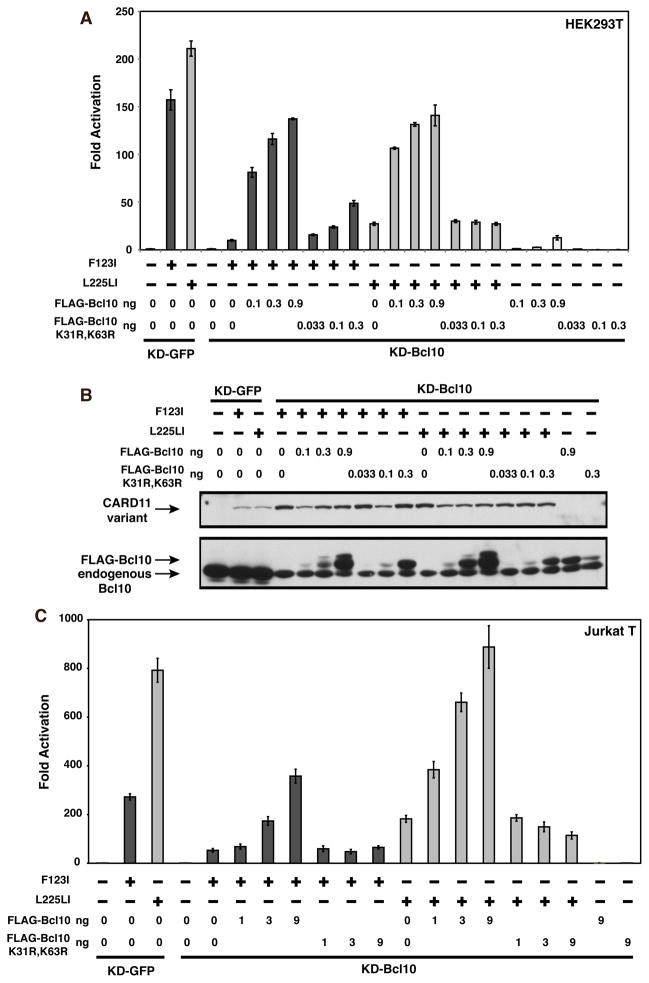
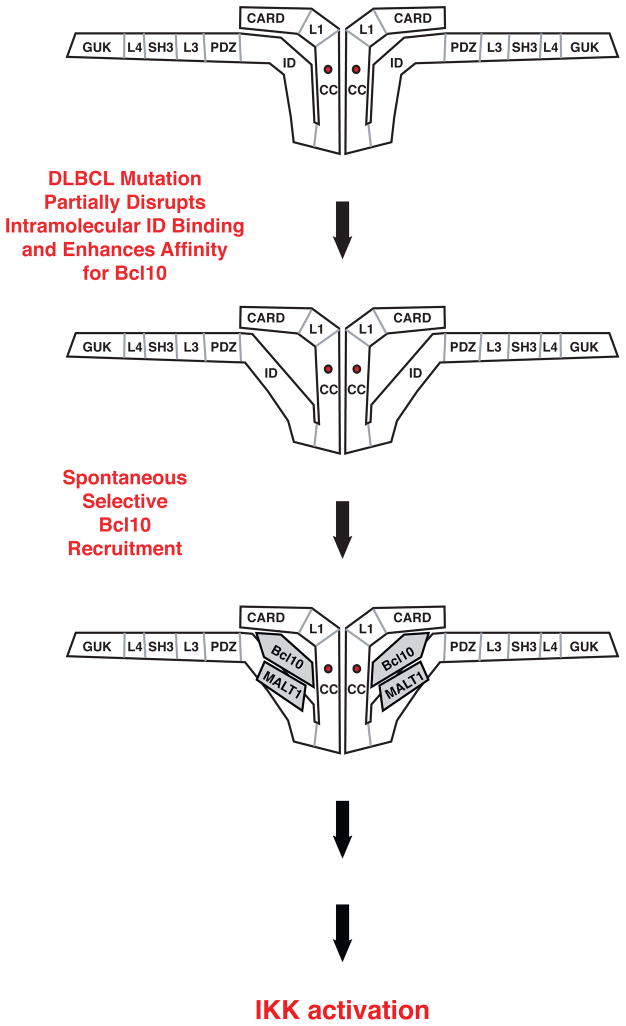
The regulated activation of NF-κB by antigen receptor signaling is required for normal B and T lymphocyte activation during the adaptive immune response. Dysregulated NF-κB activation is associated with several types of lymphoma, including diffuse large B cell lymphoma (DLBCL). During normal antigen receptor signaling, the multidomain scaffold protein CARD11 undergoes a transition from a closed, inactive state to an open, active conformation that recruits several signaling proteins into a complex, leading to IKK kinase activation. This transition is regulated by the CARD11 inhibitory domain (ID), which participates in intramolecular interactions that prevent cofactor binding to CARD11 prior to signaling, but which is neutralized after receptor engagement by phosphorylation. Several oncogenic CARD11 mutations have been identified in DLBCL that enhance activity and that are mostly found in the coiled-coil domain. However, the mechanisms by which these mutations cause CARD11 hyperactivity and spontaneous NF-κB activation are poorly understood. In this report, we provide several lines of evidence that oncogenic mutations F123I and L225LI induce CARD11 hyperactivity by disrupting autoinhibition by the CARD11 ID. These mutations disrupt ID-mediated intramolecular interactions and ID-dependent inhibition and bypass the requirement for ID phosphorylation during T cell receptor signaling. Intriguingly, these mutations selectively enhance the apparent affinity of CARD11 for Bcl10, but not for other signaling proteins that are recruited to CARD11 in an ID-dependent manner during normal antigen receptor signaling. Our results establish a mechanism that explains how DLBCL-associated mutations in CARD11 can initiate spontaneous, receptor-independent activation of NF-κB.
Figures
References
-
- Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor kappa B. Immunity. 2006;25:701–715. - PubMed
-
- Thome M, Weil R. Post-translational modifications regulate distinct functions of CARMA1 and BCL10. Trends Immunol. 2007;28:281–288. - PubMed
-
- Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol. 2006;6:799–812. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
