

Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy
- PMID: 20801068
- PMCID: PMC2991562
- DOI: 10.1016/j.ymgme.2010.08.001
Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy
Abstract
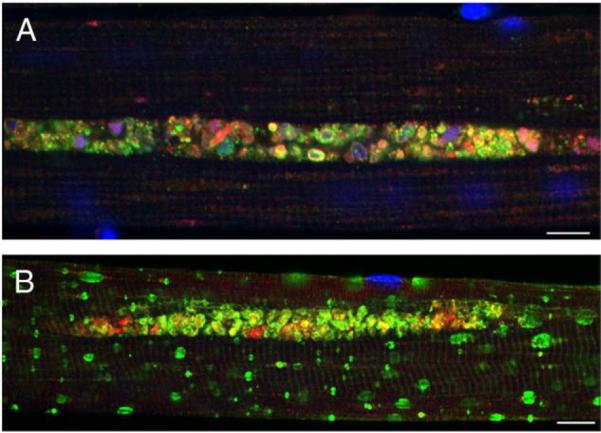
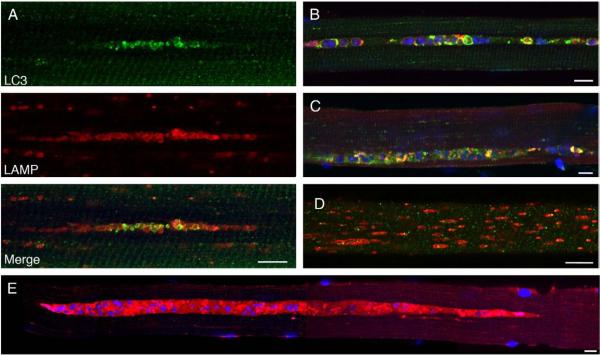
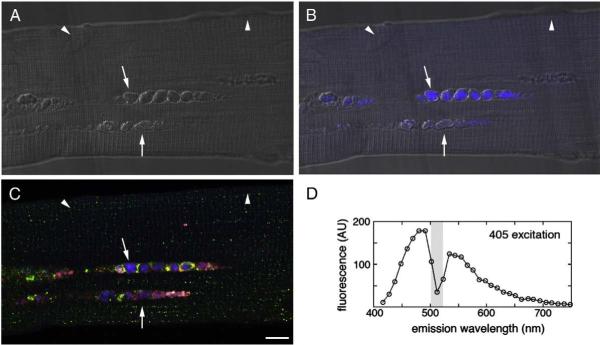
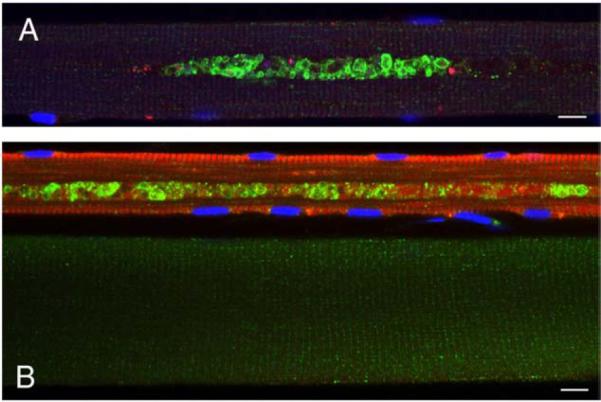
Pompe disease is a lysosomal storage disorder caused by the deficiency of acid alpha-glucosidase, the enzyme that degrades glycogen in the lysosomes. The disease manifests as a fatal cardiomyopathy and skeletal muscle myopathy in infants; in milder late-onset forms skeletal muscle is the major tissue affected. We have previously demonstrated that autophagic inclusions in muscle are prominent in adult patients and the mouse model. In this study we have evaluated the contribution of the autophagic pathology in infants before and 6 months after enzyme replacement therapy. Single muscle fibers, isolated from muscle biopsies, were stained for autophagosomal and lysosomal markers and analyzed by confocal microscopy. In addition, unstained bundles of fixed muscles were analyzed by second harmonic imaging. Unexpectedly, the autophagic component which is so prominent in juvenile and adult patients was negligible in infants; instead, the overwhelming characteristic was the presence of hugely expanded lysosomes. After 6 months on therapy, however, the autophagic buildup becomes visible as if unmasked by the clearance of glycogen. In most fibers, the two pathologies did not seem to coexist. These data point to the possibility of differences in the pathogenesis of Pompe disease in infants and adults.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
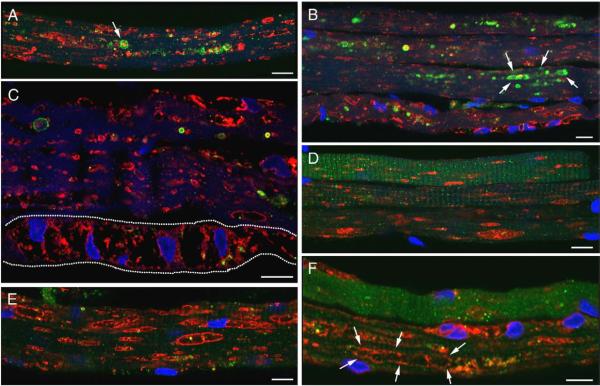
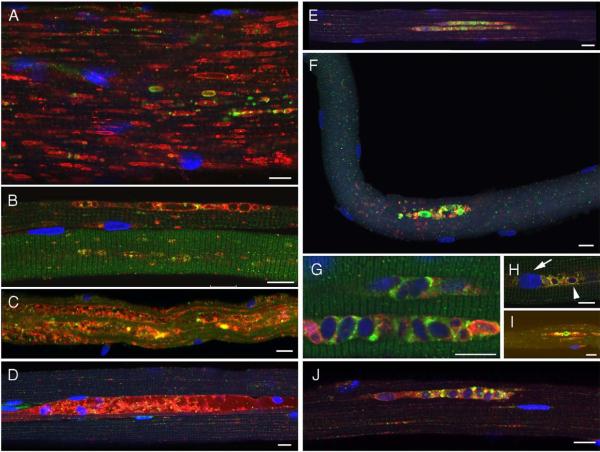
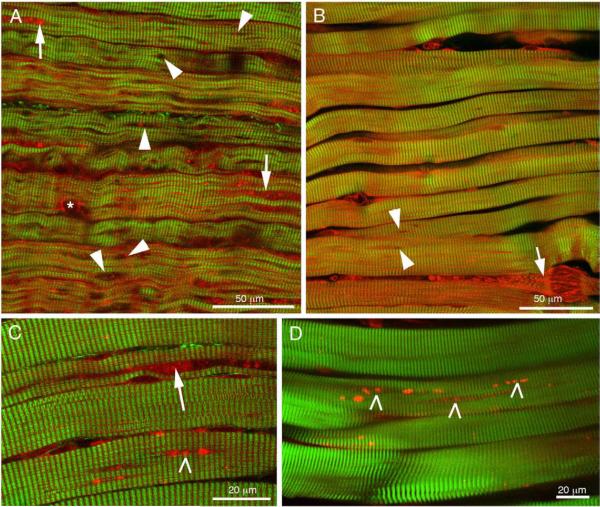
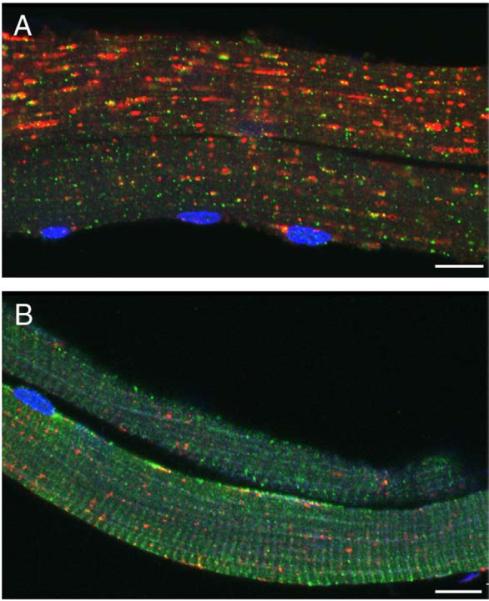
References
-
- Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. - PubMed
-
- Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York, NY: 2001. pp. 3389–3420.
-
- Engel AG, Hirschhorn R, Huie ML. Acid Maltase Deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York, NY: 2003. pp. 1559–1586.
-
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006;148:671–676. - PubMed
-
- Winkel LP, Hagemans ML, Van Doorn PA, Loonen MC, Hop WJ, Reuser AJ, Van der Ploeg AT. The natural course of non-classic Pompe's disease; a review of 225 published cases. J. Neurol. 2005;252:875–884. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
