

Mitotic phosphorylation of Cdc25B Ser321 disrupts 14-3-3 binding to the high affinity Ser323 site
- PMID: 20801879
- PMCID: PMC2966050
- DOI: 10.1074/jbc.M110.138412
Mitotic phosphorylation of Cdc25B Ser321 disrupts 14-3-3 binding to the high affinity Ser323 site
Abstract
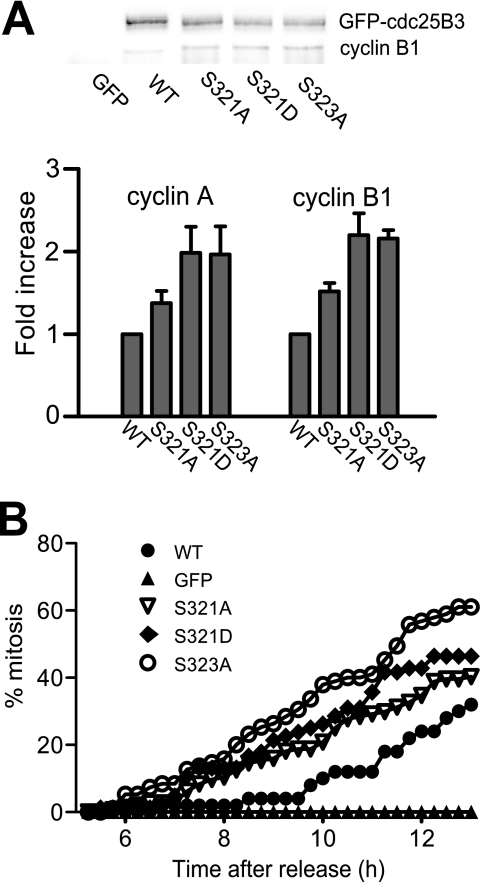
Cdc25B is a key regulator of entry into mitosis, and its activity and localization are regulated by binding of the 14-3-3 dimer. There are three 14-3-3 binding sites on Cdc25B, with Ser(323) being the highest affinity binding and is highly homologous to the Ser(216) 14-3-3 binding site on Cdc25C. Loss of 14-3-3 binding to Ser(323) increases cyclin/Cdk substrate access to the catalytic site, thereby increasing its activity. It also affects the localization of Cdc25B. Thus, phosphorylation and 14-3-3 binding to this site is essential for down-regulating Cdc25B activity, blocking its mitosis promoting function. The question of how this inhibitory signal is relieved to allow Cdc25B activation and entry into mitosis is yet to be resolved. Here, we show that Ser(323) phosphorylation is maintained into mitosis, but phosphorylation of Ser(321) disrupts 14-3-3 binding to Ser(323), mimicking the effect of inhibiting Ser(323) phosphorylation on both Cdc25B activity and localization. The unphosphorylated Ser(321) appears to have a role in stabilizing 14-3-3 binding to Ser(323), and loss of the Ser hydroxyl group appears to be sufficient to significantly reduce 14-3-3 binding. A consequence of loss of 14-3-3 binding is dephosphorylation of Ser(323). Ser(321) is phosphorylated in mitosis by Cdk1. The mitotic phosphorylation of Ser(321) acts to maintain full activation of Cdc25B by disrupting 14-3-3 binding to Ser(323) and enhancing the dephosphorylation of Ser(323) to block 14-3-3 binding to this site.
Figures


References
-
- Gabrielli B. G., De Souza C. P., Tonks I. D., Clark J. M., Hayward N. K., Ellem K. A. (1996) J. Cell Sci. 109, 1081–1093 - PubMed
-
- Lammer C., Wagerer S., Saffrich R., Mertens D., Ansorge W., Hoffmann I. (1998) J. Cell Sci. 111, 2445–2453 - PubMed
-
- Goldstone S., Pavey S., Forrest A., Sinnamon J., Gabrielli B. (2001) Oncogene 20, 921–932 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
