

Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage
- PMID: 20802089
- PMCID: PMC2945193
- DOI: 10.1101/gr.110114.110
Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage
Abstract
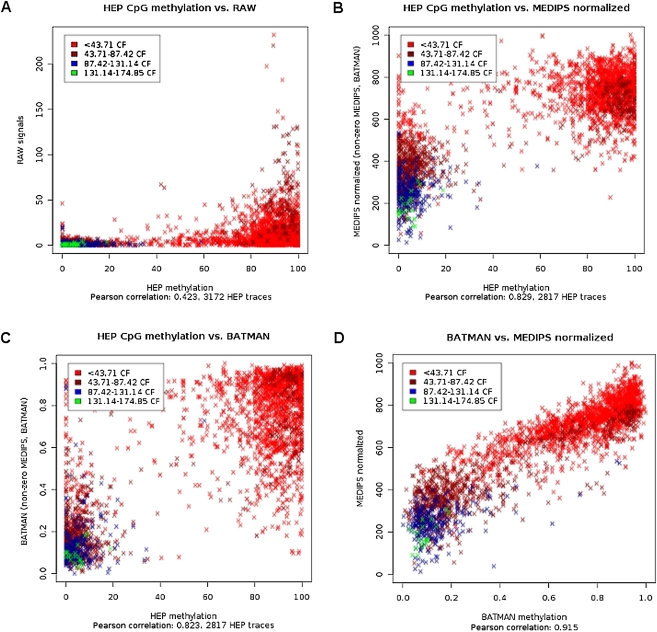
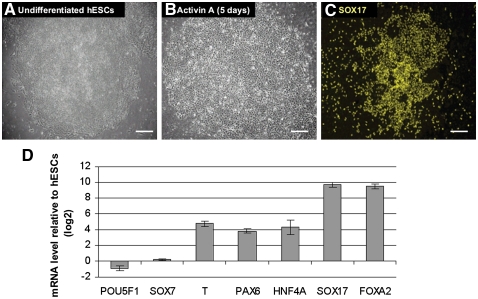
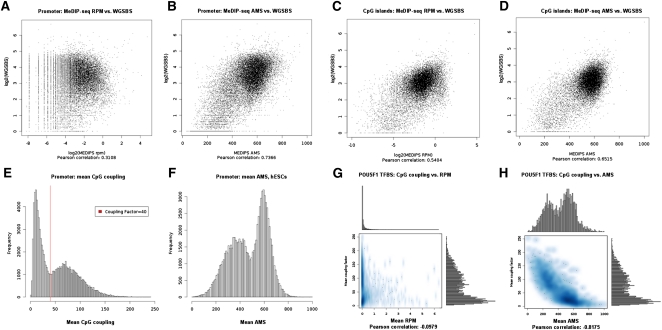
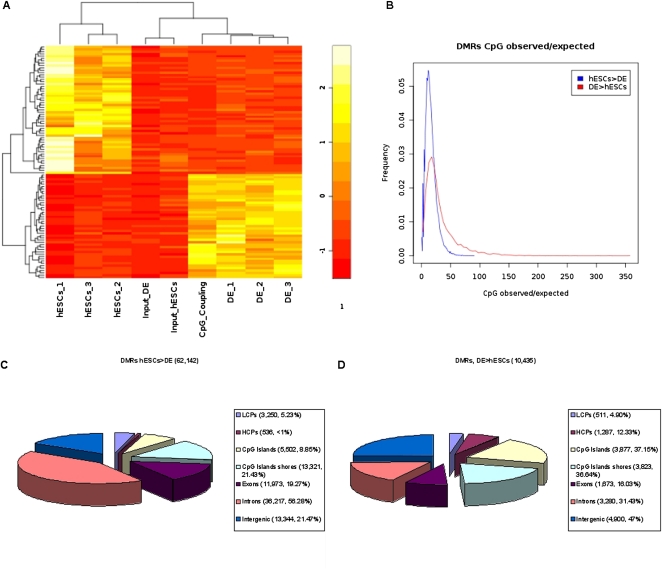
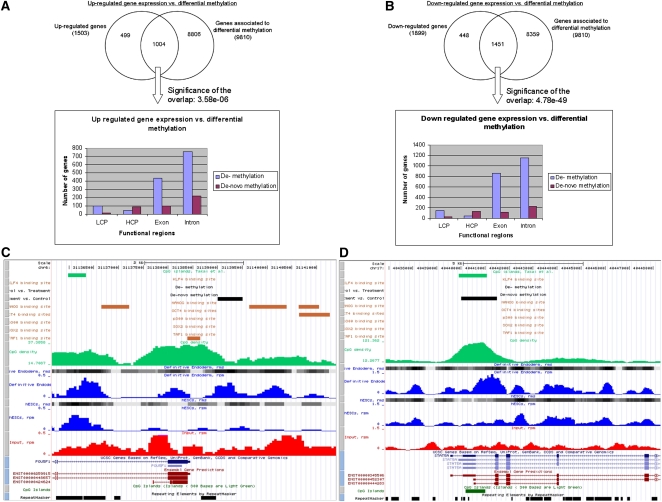
The generation of genome-wide data derived from methylated DNA immunoprecipitation followed by sequencing (MeDIP-seq) has become a major tool for epigenetic studies in health and disease. The computational analysis of such data, however, still falls short on accuracy, sensitivity, and speed. We propose a time-efficient statistical method that is able to cope with the inherent complexity of MeDIP-seq data with similar performance compared with existing methods. In order to demonstrate the computational approach, we have analyzed alterations in DNA methylation during the differentiation of human embryonic stem cells (hESCs) to definitive endoderm. We show improved correlation of normalized MeDIP-seq data in comparison to available whole-genome bisulfite sequencing data, and investigated the effect of differential methylation on gene expression. Furthermore, we analyzed the interplay between DNA methylation, histone modifications, and transcription factor binding and show that in contrast to de novo methylation, demethylation is mainly associated with regions of low CpG densities.
Figures
References
-
- Agarwal S, Holton KL, Lanza R 2008. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells 26: 1117–1127 - PubMed
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 - PubMed
Publication types
MeSH terms
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases