

Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining
- PMID: 20802485
- PMCID: PMC3018776
- DOI: 10.1038/nsmb.1899
Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining
Abstract
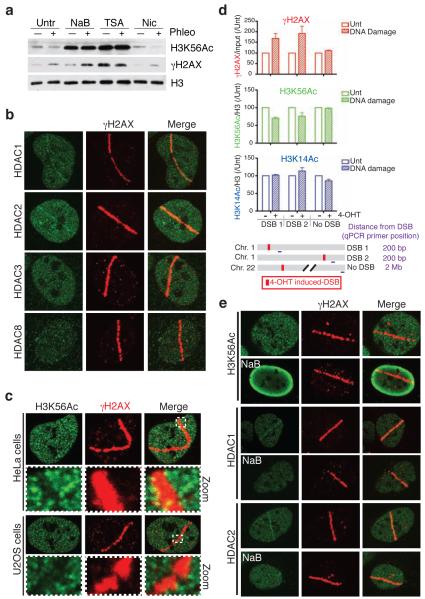
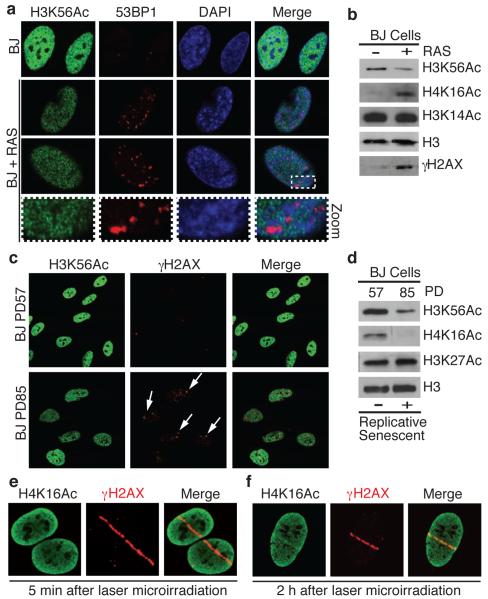
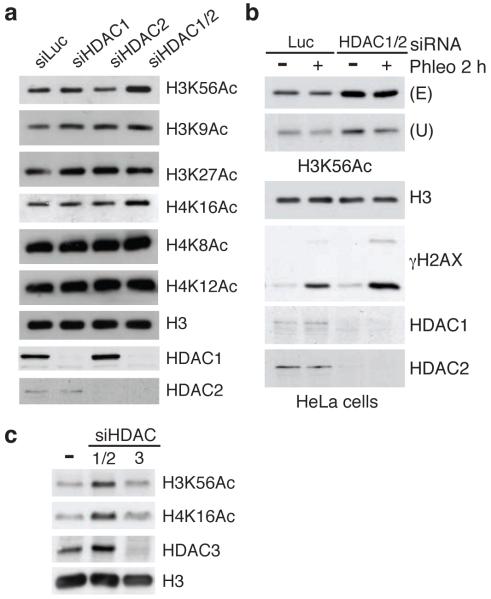
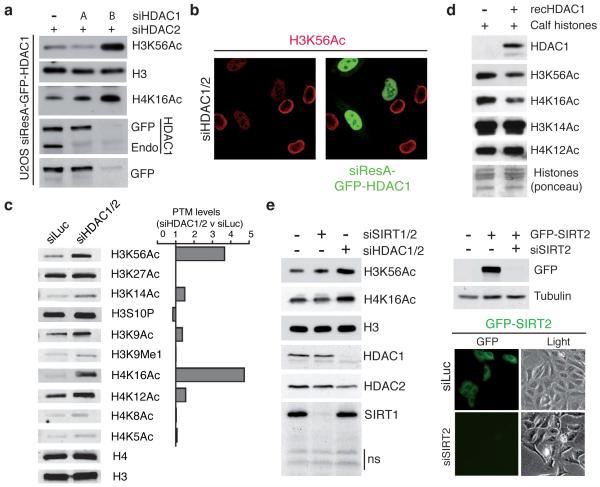
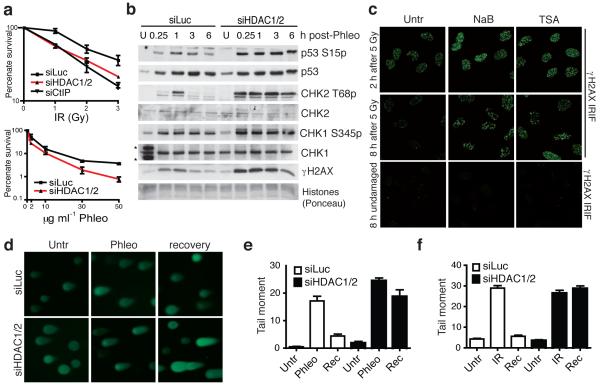
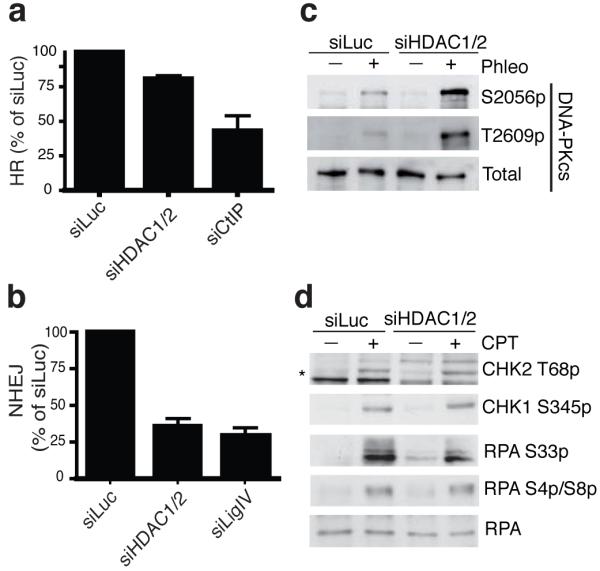
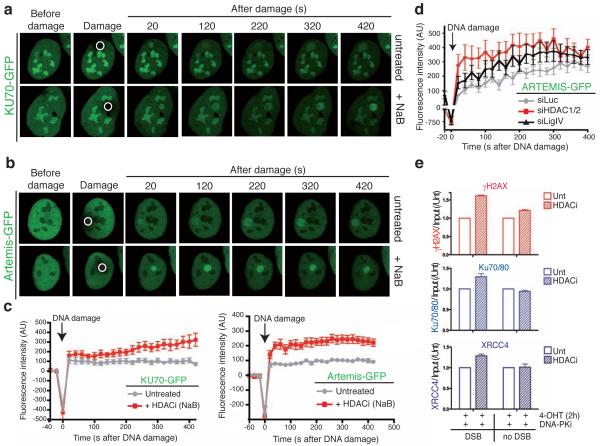
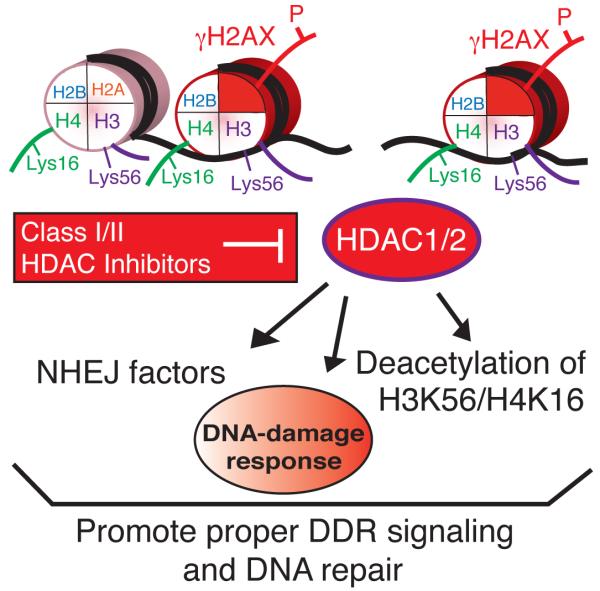
DNA double-strand break (DSB) repair occurs within chromatin and can be modulated by chromatin-modifying enzymes. Here we identify the related human histone deacetylases HDAC1 and HDAC2 as two participants in the DNA-damage response. We show that acetylation of histone H3 Lys56 (H3K56) was regulated by HDAC1 and HDAC2 and that HDAC1 and HDAC2 were rapidly recruited to DNA-damage sites to promote hypoacetylation of H3K56. Furthermore, HDAC1- and 2-depleted cells were hypersensitive to DNA-damaging agents and showed sustained DNA-damage signaling, phenotypes that reflect defective DSB repair, particularly by nonhomologous end-joining (NHEJ). Collectively, these results show that HDAC1 and HDAC2 function in the DNA-damage response by promoting DSB repair and thus provide important insights into the radio-sensitizing effects of HDAC inhibitors that are being developed as cancer therapies.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
