

Loss-of-function variants in the genomes of healthy humans
- PMID: 20805107
- PMCID: PMC2953739
- DOI: 10.1093/hmg/ddq365
Loss-of-function variants in the genomes of healthy humans
Abstract
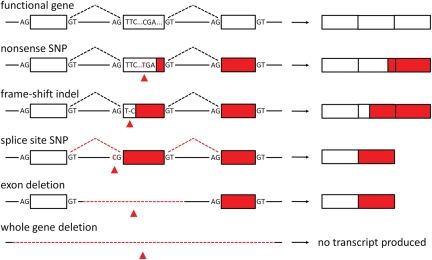
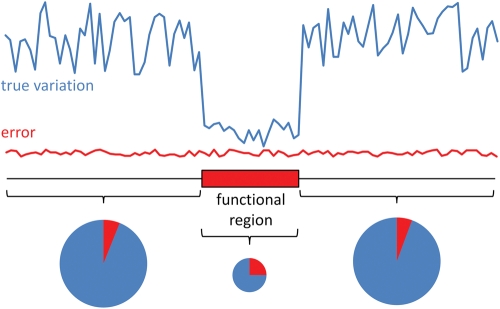
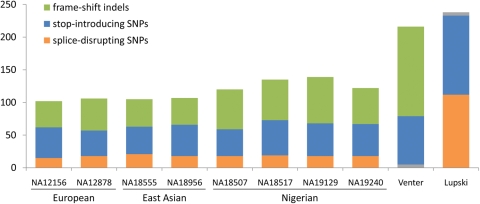
Genetic variants predicted to seriously disrupt the function of human protein-coding genes-so-called loss-of-function (LOF) variants-have traditionally been viewed in the context of severe Mendelian disease. However, recent large-scale sequencing and genotyping projects have revealed a surprisingly large number of these variants in the genomes of apparently healthy individuals--at least 100 per genome, including more than 30 in a homozygous state--suggesting a previously unappreciated level of variation in functional gene content between humans. These variants are mostly found at low frequency, suggesting that they are enriched for mildly deleterious polymorphisms suppressed by negative natural selection, and thus represent an attractive set of candidate variants for complex disease susceptibility. However, they are also enriched for sequencing and annotation artefacts, so overall present serious challenges for clinical sequencing projects seeking to identify severe disease genes amidst the 'noise' of technical error and benign genetic polymorphism. Systematic, high-quality catalogues of LOF variants present in the genomes of healthy individuals, built from the output of large-scale sequencing studies such as the 1000 Genomes Project, will help to distinguish between benign and disease-causing LOF variants, and will provide valuable resources for clinical genomics.
Figures
References
-
- Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A., et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet. 2010;42:30–35. doi:10.1038/ng.499. - DOI - PMC - PubMed
-
- Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi:10.1038/nature08516. - DOI - PMC - PubMed
-
- Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi:10.1038/nature05329. - DOI - PMC - PubMed
-
- Yngvadottir B., Xue Y., Searle S., Hunt S., Delgado M., Morrison J., Whittaker P., Deloukas P., Tyler-Smith C. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am. J. Hum. Genet. 2009;84:224–234. doi:10.1016/j.ajhg.2009.01.008. - DOI - PMC - PubMed
-
- North K.N., Beggs A.H. Deficiency of a skeletal muscle isoform of alpha-actinin (alpha-actinin-3) in merosin-positive congenital muscular dystrophy. Neuromuscul. Disord. 1996;6:229–235. doi:10.1016/0960-8966(96)00361-6. - DOI - PubMed
