

The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone
- PMID: 20805228
- PMCID: PMC2966059
- DOI: 10.1074/jbc.M110.133579
The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone
Abstract
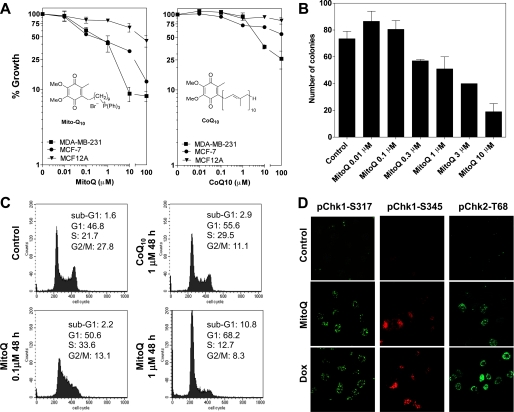
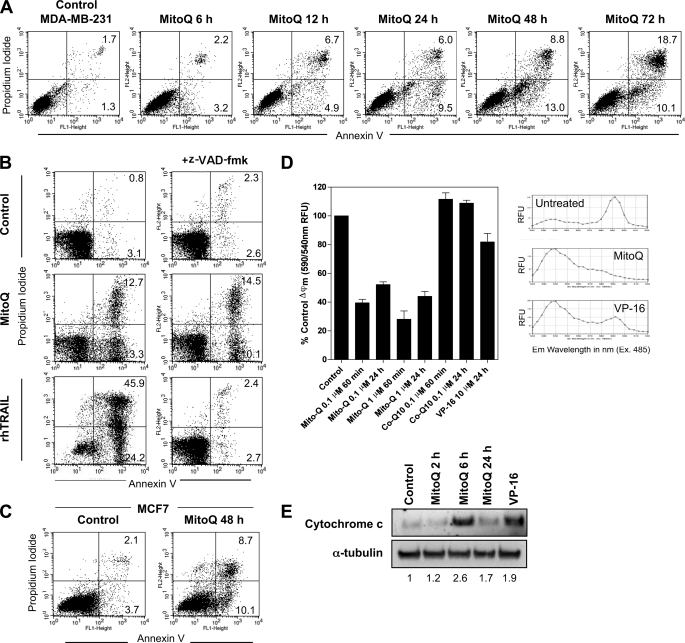
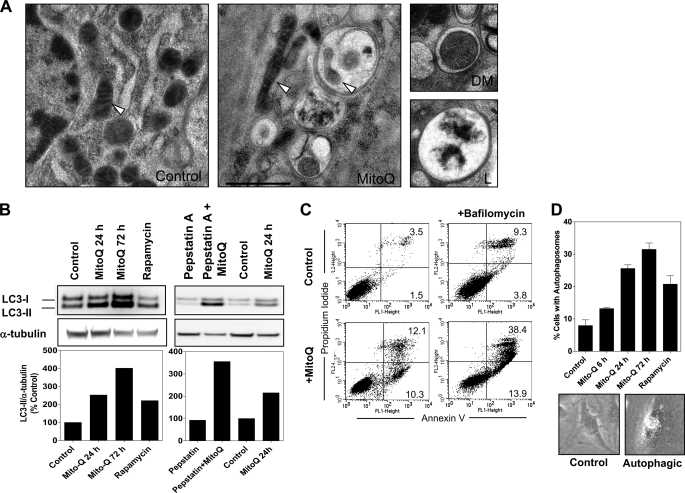
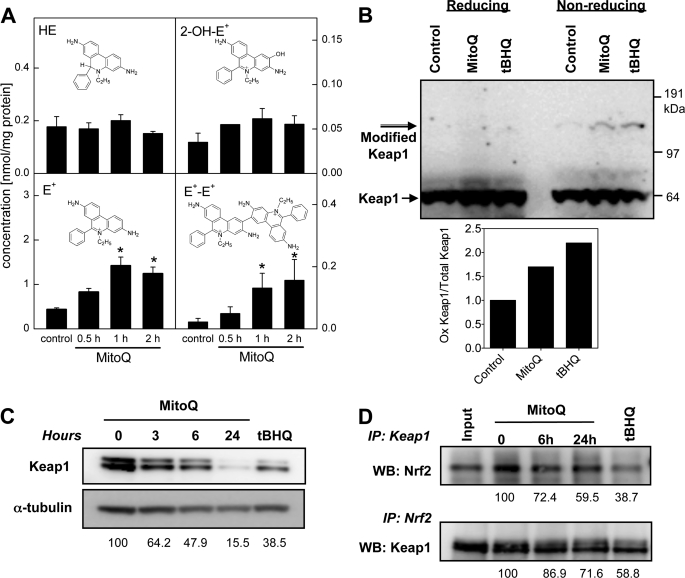
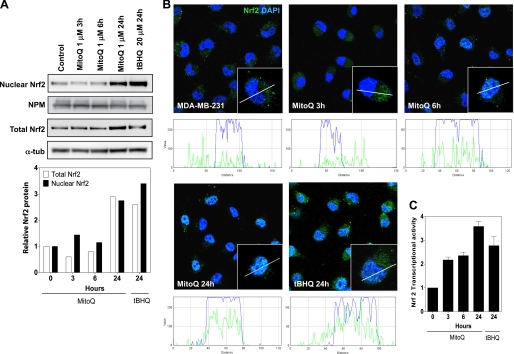
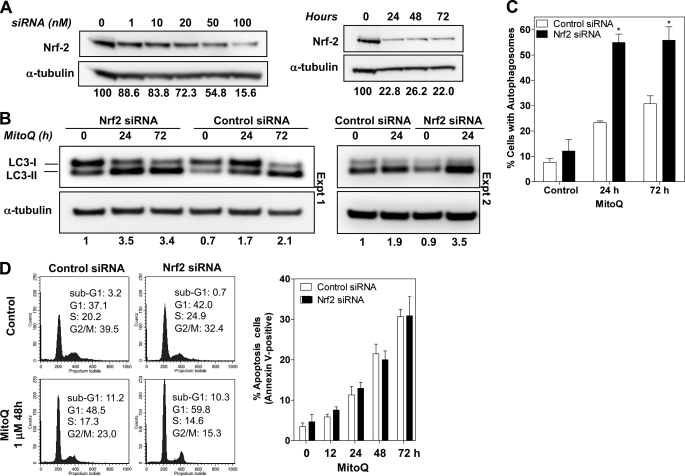
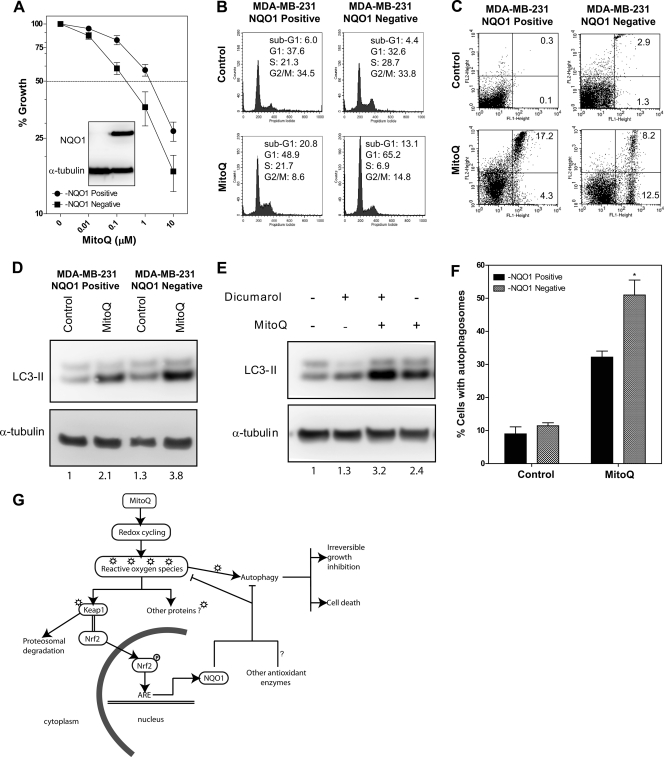
Mitoquinone (MitoQ) is a synthetically modified, redox-active ubiquinone compound that accumulates predominantly in mitochondria. We found that MitoQ is 30-fold more cytotoxic to breast cancer cells than to healthy mammary cells. MitoQ treatment led to irreversible inhibition of clonogenic growth of breast cancer cells through a combination of autophagy and apoptotic cell death mechanisms. Relatively limited cytotoxicity was seen with the parent ubiquinone coenzyme Q(10.) Inhibition of cancer cell growth by MitoQ was associated with G(1)/S cell cycle arrest and phosphorylation of the checkpoint kinases Chk1 and Chk2. The possible role of oxidative stress in MitoQ activity was investigated by measuring the products of hydroethidine oxidation. Increases in ethidium and dihydroethidium levels, markers of one-electron oxidation of hydroethidine, were observed at cytotoxic concentrations of MitoQ. Keap1, an oxidative stress sensor protein that regulates the antioxidant transcription factor Nrf2, underwent oxidation, degradation, and dissociation from Nrf2 in MitoQ-treated cells. Nrf2 protein levels, nuclear localization, and transcriptional activity also increased following MitoQ treatment. Knockdown of Nrf2 caused a 2-fold increase in autophagy and an increase in G(1) cell cycle arrest in response to MitoQ but had no apparent effect on apoptosis. The Nrf2-regulated enzyme NQO1 is partly responsible for controlling the level of autophagy. Keap1 and Nrf2 act as redox sensors for oxidative perturbations that lead to autophagy. MitoQ and similar compounds should be further evaluated for novel anticancer activity.
Figures
References
-
- Murphy M. P. (1997) Trends Biotechnol. 15, 326–330 - PubMed
-
- Murphy M. P. (2008) Biochim. Biophys. Acta 1777, 1028–1031 - PubMed
-
- Kelso G. F., Porteous C. M., Coulter C. V., Hughes G., Porteous W. K., Ledgerwood E. C., Smith R. A., Murphy M. P. (2001) J. Biol. Chem. 276, 4588–4596 - PubMed
-
- Koopman W. J., Verkaart S., Visch H. J., van der Westhuizen F. H., Murphy M. P., van den Heuvel L. W., Smeitink J. A., Willems P. H. (2005) Am. J. Physiol. Cell Physiol. 288, C1440–C1450 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
