

Natural and engineered nicking endonucleases--from cleavage mechanism to engineering of strand-specificity
- PMID: 20805246
- PMCID: PMC3017599
- DOI: 10.1093/nar/gkq742
Natural and engineered nicking endonucleases--from cleavage mechanism to engineering of strand-specificity
Erratum in
- Nucleic Acids Res. 2011 Mar 1;39(5):1966
Abstract
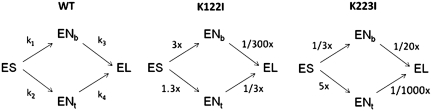
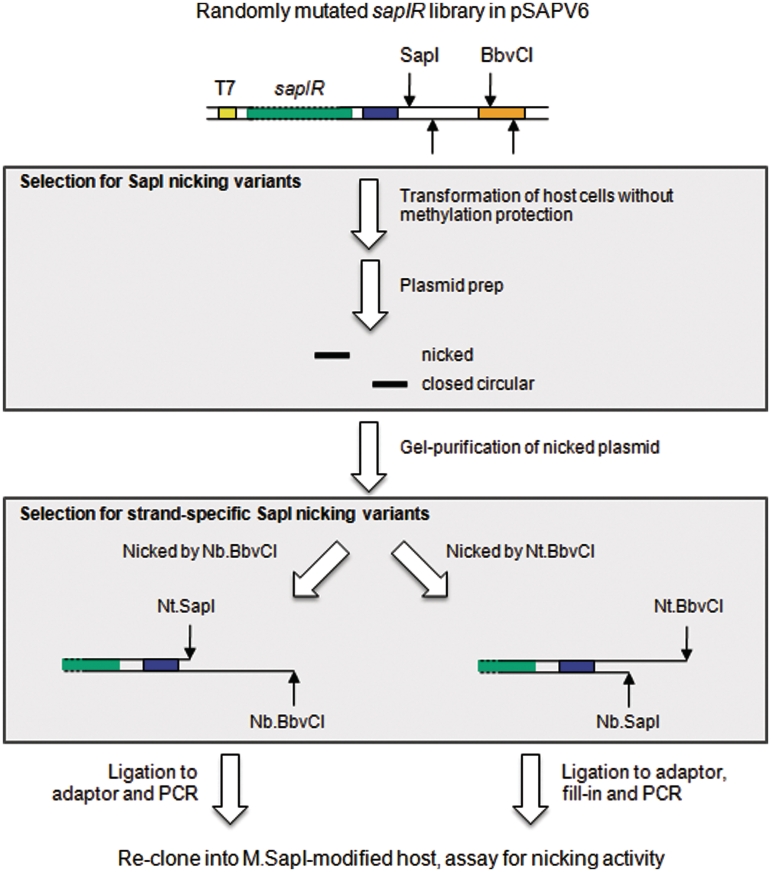
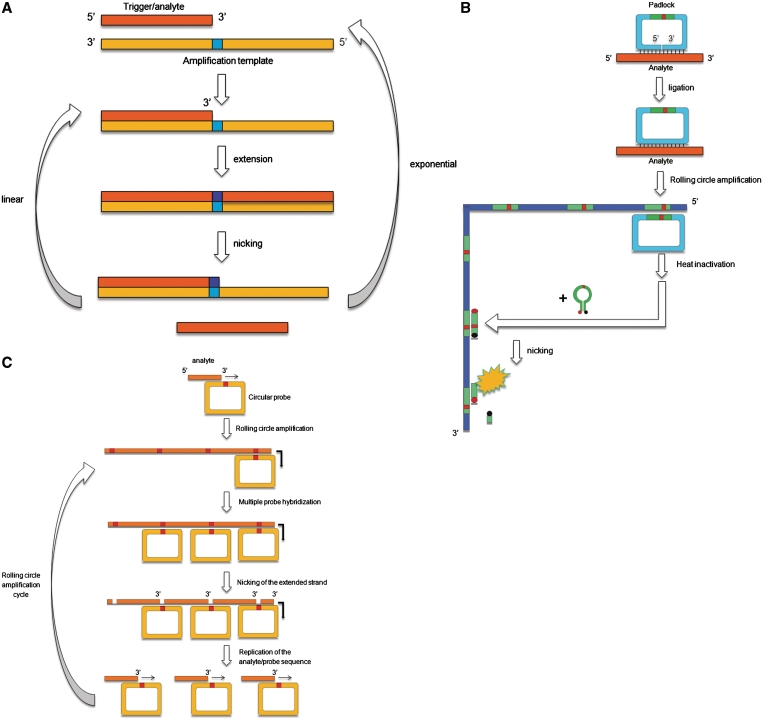
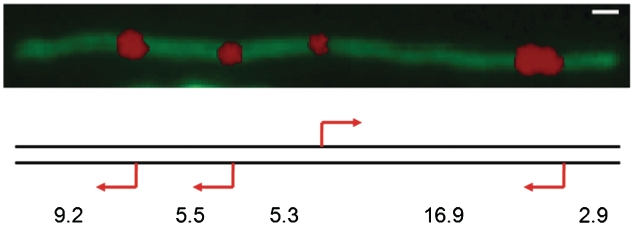
Restriction endonucleases (REases) are highly specific DNA scissors that have facilitated the development of modern molecular biology. Intensive studies of double strand (ds) cleavage activity of Type IIP REases, which recognize 4-8 bp palindromic sequences, have revealed a variety of mechanisms of molecular recognition and catalysis. Less well-studied are REases which cleave only one of the strands of dsDNA, creating a nick instead of a ds break. Naturally occurring nicking endonucleases (NEases) range from frequent cutters such as Nt.CviPII (^CCD; ^ denotes the cleavage site) to rare-cutting homing endonucleases (HEases) such as I-HmuI. In addition to these bona fida NEases, individual subunits of some heterodimeric Type IIS REases have recently been shown to be natural NEases. The discovery and characterization of more REases that recognize asymmetric sequences, particularly Types IIS and IIA REases, has revealed recognition and cleavage mechanisms drastically different from the canonical Type IIP mechanisms, and has allowed researchers to engineer highly strand-specific NEases. Monomeric LAGLIDADG HEases use two separate catalytic sites for cleavage. Exploitation of this characteristic has also resulted in useful nicking HEases. This review aims at providing an overview of the cleavage mechanisms of Types IIS and IIA REases and LAGLIDADG HEases, the engineering of their nicking variants, and the applications of NEases and nicking HEases.
Figures
Similar articles
-
I-Ssp6803I: the first homing endonuclease from the PD-(D/E)XK superfamily exhibits an unusual mode of DNA recognition.Bioinformatics. 2007 Mar 1;23(5):527-30. doi: 10.1093/bioinformatics/btm007. Epub 2007 Jan 22. Bioinformatics. 2007. PMID: 17242028
-
Engineering Infrequent DNA Nicking Endonuclease by Fusion of a BamHI Cleavage-Deficient Mutant and a DNA Nicking Domain.Front Microbiol. 2022 Feb 1;12:787073. doi: 10.3389/fmicb.2021.787073. eCollection 2021. Front Microbiol. 2022. PMID: 35178039 Free PMC article.
-
Engineering strand-specific DNA nicking enzymes from the type IIS restriction endonucleases BsaI, BsmBI, and BsmAI.J Mol Biol. 2004 Mar 26;337(3):573-83. doi: 10.1016/j.jmb.2004.02.003. J Mol Biol. 2004. PMID: 15019778
-
Sequence-specific DNA nicking endonucleases.Biomol Concepts. 2015 Aug;6(4):253-67. doi: 10.1515/bmc-2015-0016. Biomol Concepts. 2015. PMID: 26352356 Review.
-
Type II restriction endonucleases--a historical perspective and more.Nucleic Acids Res. 2014 Jul;42(12):7489-527. doi: 10.1093/nar/gku447. Epub 2014 May 30. Nucleic Acids Res. 2014. PMID: 24878924 Free PMC article. Review.
Cited by
-
Site- and strand-specific nicking of DNA by fusion proteins derived from MutH and I-SceI or TALE repeats.Nucleic Acids Res. 2013 Apr;41(7):e83. doi: 10.1093/nar/gkt080. Epub 2013 Feb 13. Nucleic Acids Res. 2013. PMID: 23408850 Free PMC article.
-
Strong subunit coordination drives a powerful viral DNA packaging motor.Proc Natl Acad Sci U S A. 2013 Apr 9;110(15):5909-14. doi: 10.1073/pnas.1222820110. Epub 2013 Mar 25. Proc Natl Acad Sci U S A. 2013. PMID: 23530228 Free PMC article.
-
Types and Applications of Nicking Enzyme-Combined Isothermal Amplification.Int J Mol Sci. 2022 Apr 21;23(9):4620. doi: 10.3390/ijms23094620. Int J Mol Sci. 2022. PMID: 35563012 Free PMC article. Review.
-
Performance of the Cas9 nickase system in Drosophila melanogaster.G3 (Bethesda). 2014 Aug 15;4(10):1955-62. doi: 10.1534/g3.114.013821. G3 (Bethesda). 2014. PMID: 25128437 Free PMC article.
-
Vectors for ligation-independent construction of lacZ gene fusions and cloning of PCR products using a nicking endonuclease.Plasmid. 2011 Sep;66(3):180-5. doi: 10.1016/j.plasmid.2011.07.007. Epub 2011 Aug 10. Plasmid. 2011. PMID: 21854804 Free PMC article.
References
-
- Zilberman D, Henikoff S. Genome-wide analysis of DNA methylation patterns. Development. 2007;134:3959–3965. - PubMed
-
- Nielsen KL. DeepSAGE: higher sensitivity and multiplexing of samples using a simpler experimental protocol. Methods Mol. Biol. 2008;387:81–94. - PubMed
-
- Porter D, Yao J, Polyak K. SAGE and related approaches for cancer target identification. Drug Discov. Today. 2006;11:110–118. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
