

Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology
- PMID: 20805560
- PMCID: PMC2928417
- DOI: 10.2353/jmoldx.2010.100043
Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology
Abstract
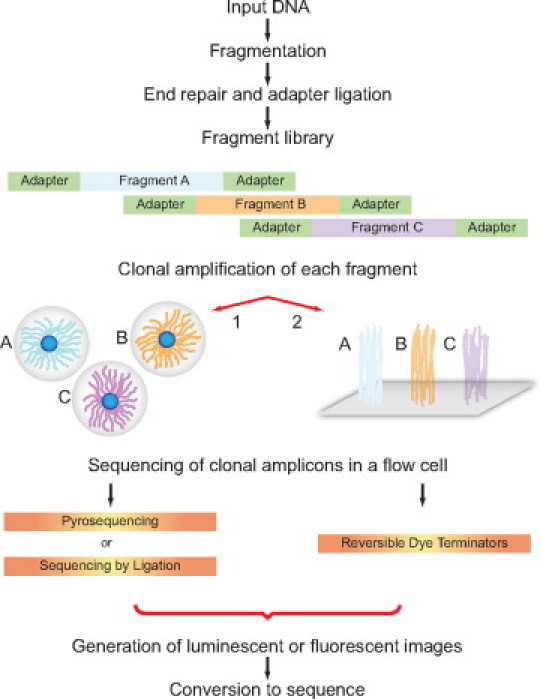
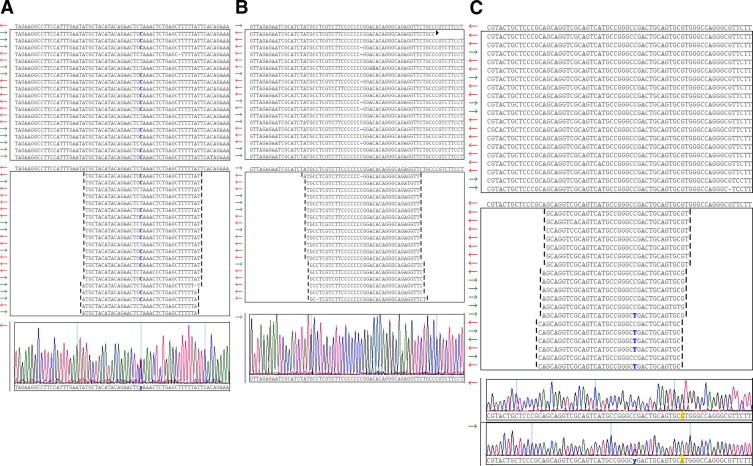
During the past five years, new high-throughput DNA sequencing technologies have emerged; these technologies are collectively referred to as next generation sequencing (NGS). By virtue of sequencing clonally amplified DNA templates or single DNA molecules in a massively parallel fashion in a flow cell, NGS provides both qualitative and quantitative sequence data. This combination of information has made NGS the technology of choice for complex genetic analyses that were previously either technically infeasible or cost prohibitive. As a result, NGS has had a fundamental and broad impact on many facets of biomedical research. In contrast, the dissemination of NGS into the clinical diagnostic realm is in its early stages. Though NGS is powerful and can be envisioned to have multiple applications in clinical diagnostics, the technology is currently complex. Successful adoption of NGS into the clinical laboratory will require expertise in both molecular biology techniques and bioinformatics. The current report presents principles that underlie NGS including sequencing library preparation, sequencing chemistries, and an introduction to NGS data analysis. These concepts are subsequently further illustrated by showing representative results from a case study using NGS for targeted resequencing of genes implicated in hypertrophic cardiomyopathy.
Figures
Similar articles
-
Next-generation sequencing: from basic research to diagnostics.Clin Chem. 2009 Apr;55(4):641-58. doi: 10.1373/clinchem.2008.112789. Epub 2009 Feb 26. Clin Chem. 2009. PMID: 19246620 Review.
-
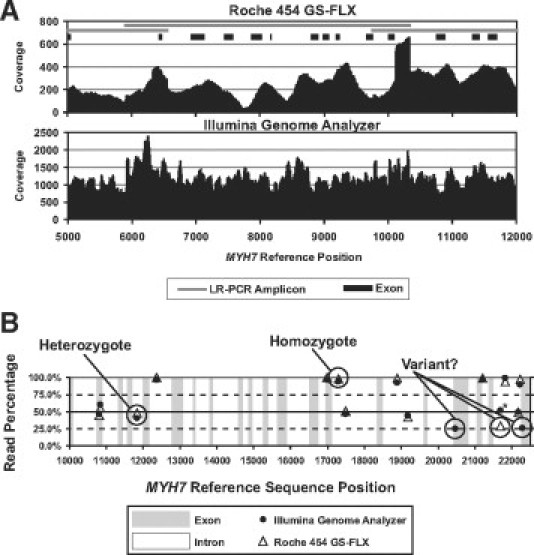
Comparison of the Illumina Genome Analyzer and Roche 454 GS FLX for resequencing of hypertrophic cardiomyopathy-associated genes.J Biomol Tech. 2010 Jul;21(2):73-80. J Biomol Tech. 2010. PMID: 20592870 Free PMC article.
-
A Clinician's Guide to Bioinformatics for Next-Generation Sequencing.J Thorac Oncol. 2023 Feb;18(2):143-157. doi: 10.1016/j.jtho.2022.11.006. Epub 2022 Nov 12. J Thorac Oncol. 2023. PMID: 36379355 Free PMC article. Review.
-
Newly designed 11-gene panel reveals first case of hereditary amyloidosis captured by massive parallel sequencing.J Clin Pathol. 2018 Aug;71(8):687-694. doi: 10.1136/jclinpath-2017-204978. Epub 2018 Feb 17. J Clin Pathol. 2018. PMID: 29455155 Free PMC article.
-
Multiplexed Reference Materials as Controls for Diagnostic Next-Generation Sequencing: A Pilot Investigating Applications for Hypertrophic Cardiomyopathy.J Mol Diagn. 2016 Nov;18(6):882-889. doi: 10.1016/j.jmoldx.2016.07.005. Epub 2016 Sep 15. J Mol Diagn. 2016. PMID: 27639548
Cited by
-
Next generation sequencing in cardiovascular diseases.World J Cardiol. 2012 Oct 26;4(10):288-95. doi: 10.4330/wjc.v4.i10.288. World J Cardiol. 2012. PMID: 23110245 Free PMC article.
-
Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis.Kidney Int. 2012 Jan;81(2):196-200. doi: 10.1038/ki.2011.315. Epub 2011 Sep 7. Kidney Int. 2012. PMID: 21900877 Free PMC article.
-
The promise of whole-exome sequencing in medical genetics.J Hum Genet. 2014 Jan;59(1):5-15. doi: 10.1038/jhg.2013.114. Epub 2013 Nov 7. J Hum Genet. 2014. PMID: 24196381 Review.
-
Exome sequencing: new insights into lipoprotein disorders.Curr Cardiol Rep. 2014 Jul;16(7):507. doi: 10.1007/s11886-014-0507-2. Curr Cardiol Rep. 2014. PMID: 24893940 Review.
-
Novel sequencing-based strategies for high-throughput discovery of genetic mutations underlying inherited antibody deficiency disorders.Curr Allergy Asthma Rep. 2011 Oct;11(5):352-60. doi: 10.1007/s11882-011-0211-x. Curr Allergy Asthma Rep. 2011. PMID: 21792638 Free PMC article. Review.
References
-
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. - PubMed
-
- Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. - PubMed
-
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. - PubMed
-
- Yeager M, Xiao N, Hayes RB, Bouffard P, Desany B, Burdett L, Orr N, Matthews C, Qi L, Crenshaw A, Markovic Z, Fredrikson KM, Jacobs KB, Amundadottir L, Jarvie TP, Hunter DJ, Hoover R, Thomas G, Harkins TT, Chanock SJ. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum Genet. 2008;124:161–170. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
