

Cancer biomarker discovery: the entropic hallmark
- PMID: 20805891
- PMCID: PMC2923618
- DOI: 10.1371/journal.pone.0012262
Cancer biomarker discovery: the entropic hallmark
Abstract
Background: It is a commonly accepted belief that cancer cells modify their transcriptional state during the progression of the disease. We propose that the progression of cancer cells towards malignant phenotypes can be efficiently tracked using high-throughput technologies that follow the gradual changes observed in the gene expression profiles by employing Shannon's mathematical theory of communication. Methods based on Information Theory can then quantify the divergence of cancer cells' transcriptional profiles from those of normally appearing cells of the originating tissues. The relevance of the proposed methods can be evaluated using microarray datasets available in the public domain but the method is in principle applicable to other high-throughput methods.
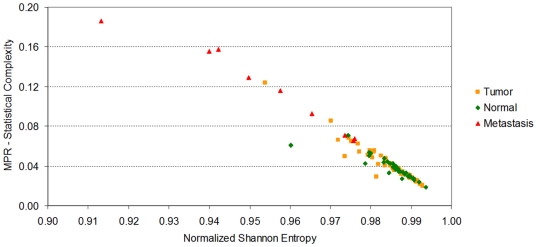
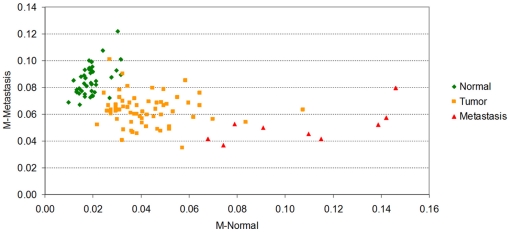
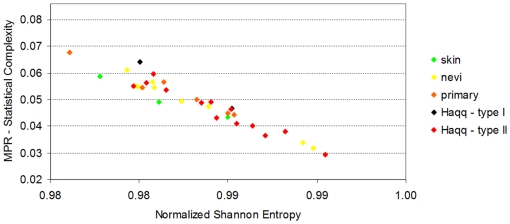
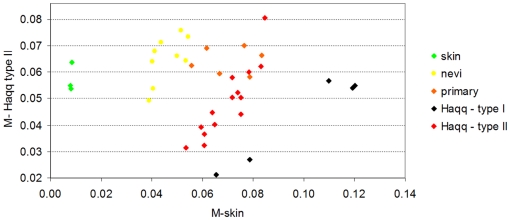
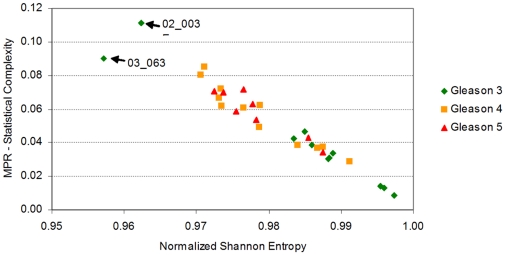
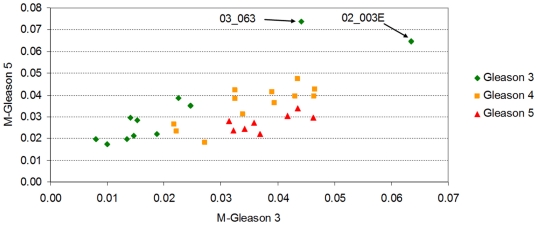
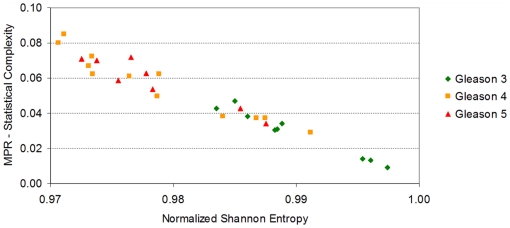
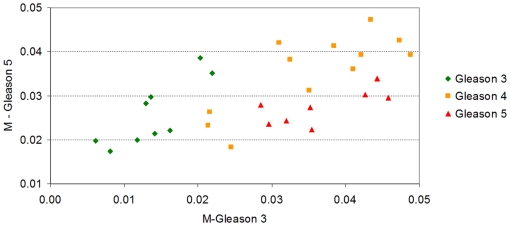
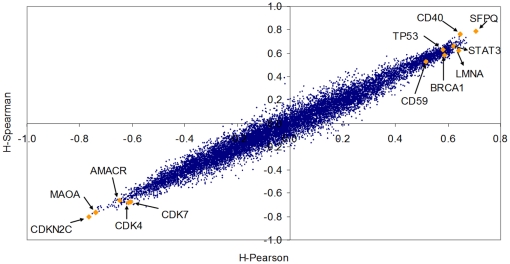
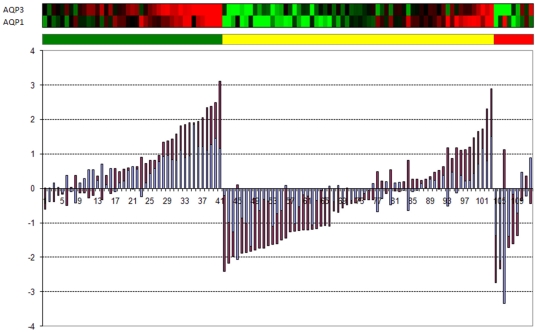
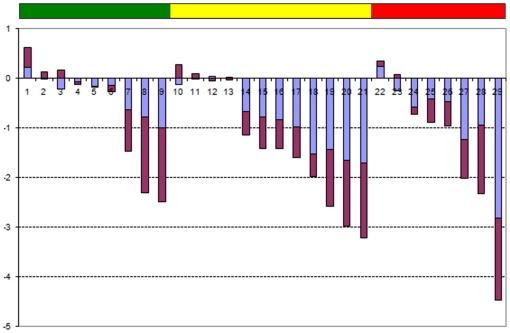
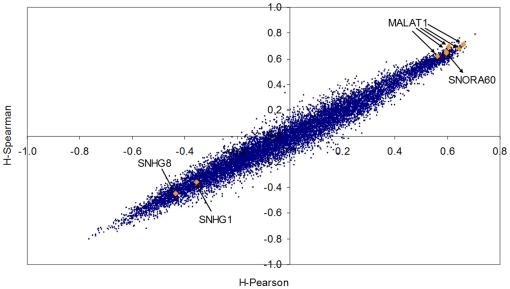
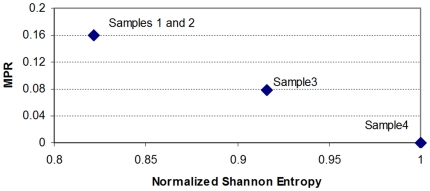
Methodology/principal findings: Using melanoma and prostate cancer datasets we illustrate how it is possible to employ Shannon Entropy and the Jensen-Shannon divergence to trace the transcriptional changes progression of the disease. We establish how the variations of these two measures correlate with established biomarkers of cancer progression. The Information Theory measures allow us to identify novel biomarkers for both progressive and relatively more sudden transcriptional changes leading to malignant phenotypes. At the same time, the methodology was able to validate a large number of genes and processes that seem to be implicated in the progression of melanoma and prostate cancer.
Conclusions/significance: We thus present a quantitative guiding rule, a new unifying hallmark of cancer: the cancer cell's transcriptome changes lead to measurable observed transitions of Normalized Shannon Entropy values (as measured by high-throughput technologies). At the same time, tumor cells increment their divergence from the normal tissue profile increasing their disorder via creation of states that we might not directly measure. This unifying hallmark allows, via the the Jensen-Shannon divergence, to identify the arrow of time of the processes from the gene expression profiles, and helps to map the phenotypical and molecular hallmarks of specific cancer subtypes. The deep mathematical basis of the approach allows us to suggest that this principle is, hopefully, of general applicability for other diseases.
Conflict of interest statement
Figures



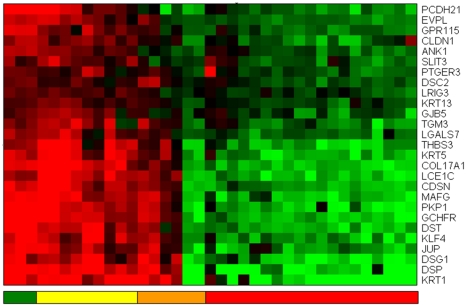
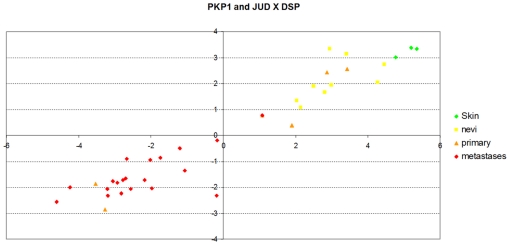
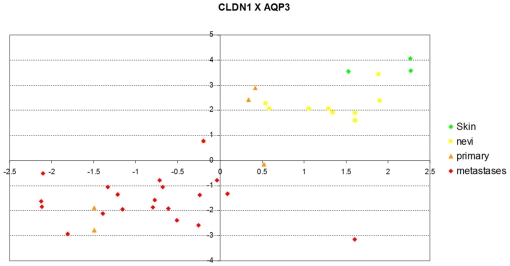
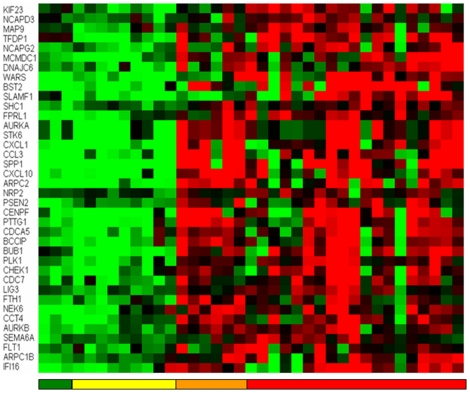


References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Wong DJ, Segal E, Chang HY. Stemness, cancer and cancer stem cells. Cell Cycle. 2008;7 - PubMed
-
- Glinsky GV. “Stemness” genomics law governs clinical behavior of human cancer: implications for decision making in disease management. J Clin Oncol. 2008;26:2846–2853. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
