

Bayesian random local clocks, or one rate to rule them all
- PMID: 20807414
- PMCID: PMC2949620
- DOI: 10.1186/1741-7007-8-114
Bayesian random local clocks, or one rate to rule them all
Abstract
Background: Relaxed molecular clock models allow divergence time dating and "relaxed phylogenetic" inference, in which a time tree is estimated in the face of unequal rates across lineages. We present a new method for relaxing the assumption of a strict molecular clock using Markov chain Monte Carlo to implement Bayesian modeling averaging over random local molecular clocks. The new method approaches the problem of rate variation among lineages by proposing a series of local molecular clocks, each extending over a subregion of the full phylogeny. Each branch in a phylogeny (subtending a clade) is a possible location for a change of rate from one local clock to a new one. Thus, including both the global molecular clock and the unconstrained model results, there are a total of 2(2n-2) possible rate models available for averaging with 1, 2, ..., 2n - 2 different rate categories.
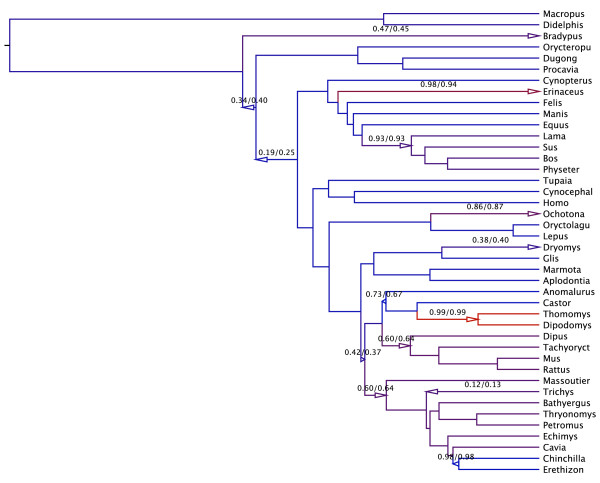
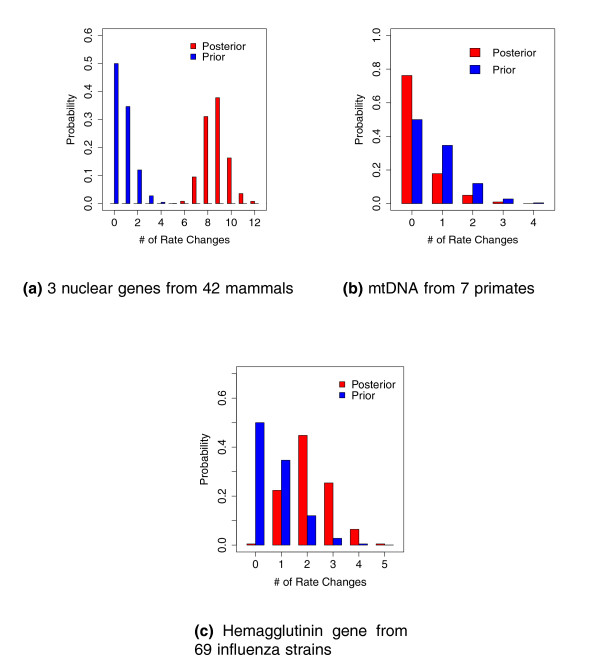
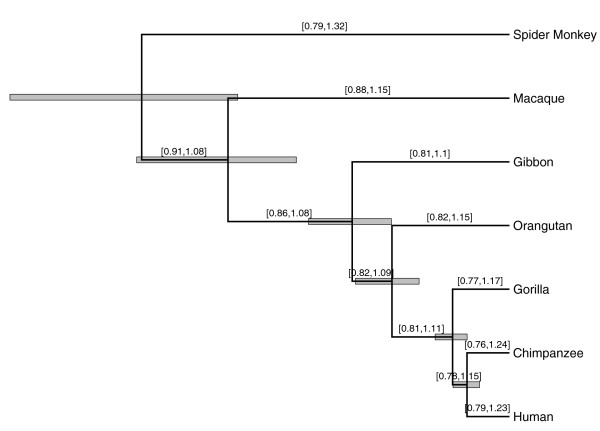
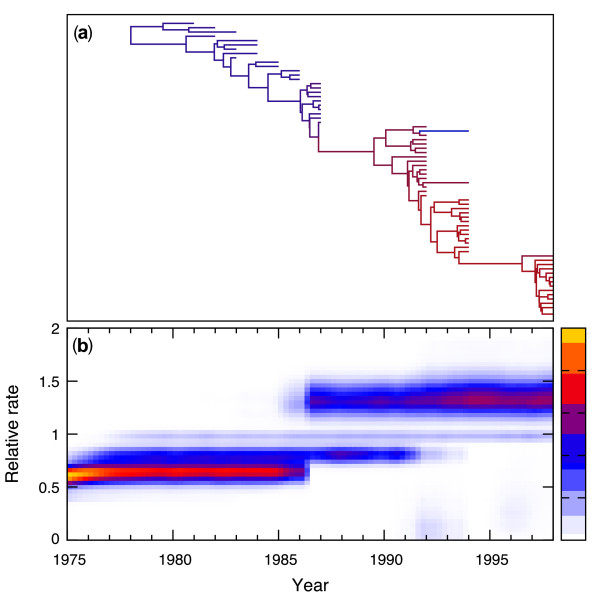
Results: We propose an efficient method to sample this model space while simultaneously estimating the phylogeny. The new method conveniently allows a direct test of the strict molecular clock, in which one rate rules them all, against a large array of alternative local molecular clock models. We illustrate the method's utility on three example data sets involving mammal, primate and influenza evolution. Finally, we explore methods to visualize the complex posterior distribution that results from inference under such models.
Conclusions: The examples suggest that large sequence datasets may only require a small number of local molecular clocks to reconcile their branch lengths with a time scale. All of the analyses described here are implemented in the open access software package BEAST 1.5.4 (http://beast-mcmc.googlecode.com/).
Figures
Similar articles
-
Model averaging and Bayes factor calculation of relaxed molecular clocks in Bayesian phylogenetics.Mol Biol Evol. 2012 Feb;29(2):751-61. doi: 10.1093/molbev/msr232. Epub 2011 Sep 22. Mol Biol Evol. 2012. PMID: 21940644 Free PMC article.
-
Shrinkage-based Random Local Clocks with Scalable Inference.Mol Biol Evol. 2023 Nov 3;40(11):msad242. doi: 10.1093/molbev/msad242. Mol Biol Evol. 2023. PMID: 37950885 Free PMC article.
-
Adaptive dating and fast proposals: Revisiting the phylogenetic relaxed clock model.PLoS Comput Biol. 2021 Feb 2;17(2):e1008322. doi: 10.1371/journal.pcbi.1008322. eCollection 2021 Feb. PLoS Comput Biol. 2021. PMID: 33529184 Free PMC article.
-
Molecular-clock methods for estimating evolutionary rates and timescales.Mol Ecol. 2014 Dec;23(24):5947-65. doi: 10.1111/mec.12953. Epub 2014 Oct 30. Mol Ecol. 2014. PMID: 25290107 Review.
-
Modeling Substitution Rate Evolution across Lineages and Relaxing the Molecular Clock.Genome Biol Evol. 2024 Sep 3;16(9):evae199. doi: 10.1093/gbe/evae199. Genome Biol Evol. 2024. PMID: 39332907 Free PMC article. Review.
Cited by
-
Avian influenza virus exhibits distinct evolutionary dynamics in wild birds and poultry.BMC Evol Biol. 2015 Jun 26;15:120. doi: 10.1186/s12862-015-0410-5. BMC Evol Biol. 2015. PMID: 26111936 Free PMC article.
-
Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene.Viruses. 2020 Jul 23;12(8):790. doi: 10.3390/v12080790. Viruses. 2020. PMID: 32717934 Free PMC article.
-
Were sea level changes during the Pleistocene in the South Atlantic Coastal Plain a driver of speciation in Petunia (Solanaceae)?BMC Evol Biol. 2015 May 20;15:92. doi: 10.1186/s12862-015-0363-8. BMC Evol Biol. 2015. PMID: 25989835 Free PMC article.
-
Heterogeneous molecular processes among the causes of how sequence similarity scores can fail to recapitulate phylogeny.Brief Bioinform. 2017 May 1;18(3):451-457. doi: 10.1093/bib/bbw034. Brief Bioinform. 2017. PMID: 27103098 Free PMC article.
-
Bayesian phylogeography finds its roots.PLoS Comput Biol. 2009 Sep;5(9):e1000520. doi: 10.1371/journal.pcbi.1000520. Epub 2009 Sep 25. PLoS Comput Biol. 2009. PMID: 19779555 Free PMC article.
References
-
- Zuckerkandl E, Pauling L. Evolutionary Divergence and Convergence in Proteins. New York: Academic Press; 1965. pp. 97–166.
-
- Thorne JL, Kishino H, Painter IS. Estimating the rate of evolution of the rate of molecular evolution. Mol Biol Evol. 1998;15:1647–1657. - PubMed
-
- Sanderson MJ. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol Biol Evol. 2002;19:101–109. - PubMed
