

Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74
- PMID: 20807568
- PMCID: PMC3586206
- DOI: 10.1016/j.cellsig.2010.08.013
Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74
Abstract
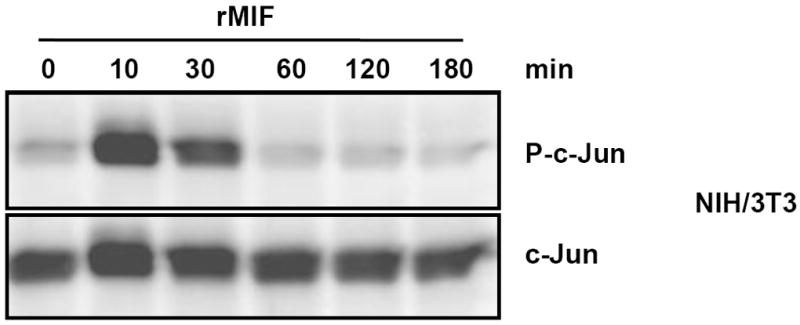
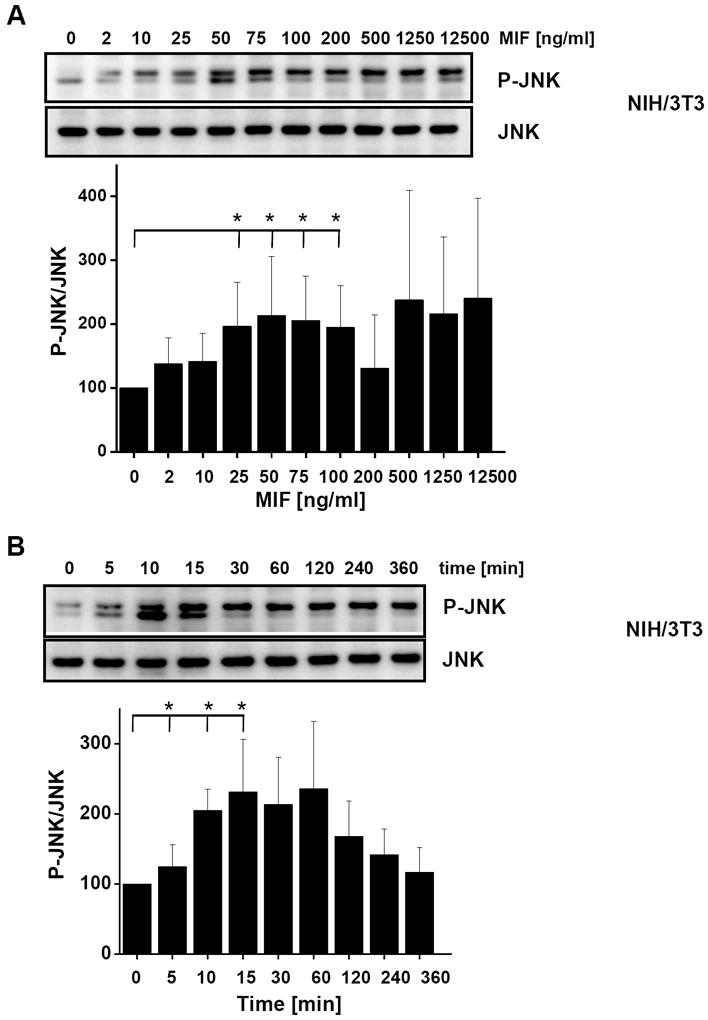
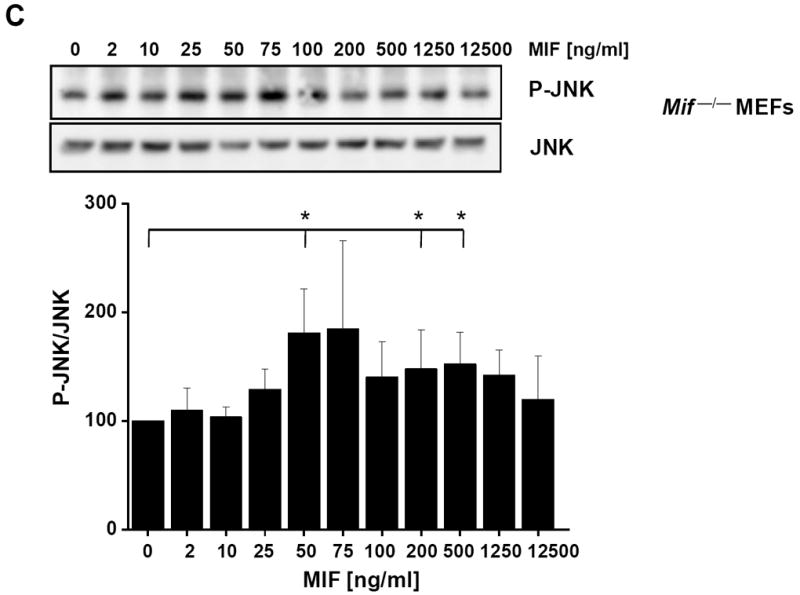
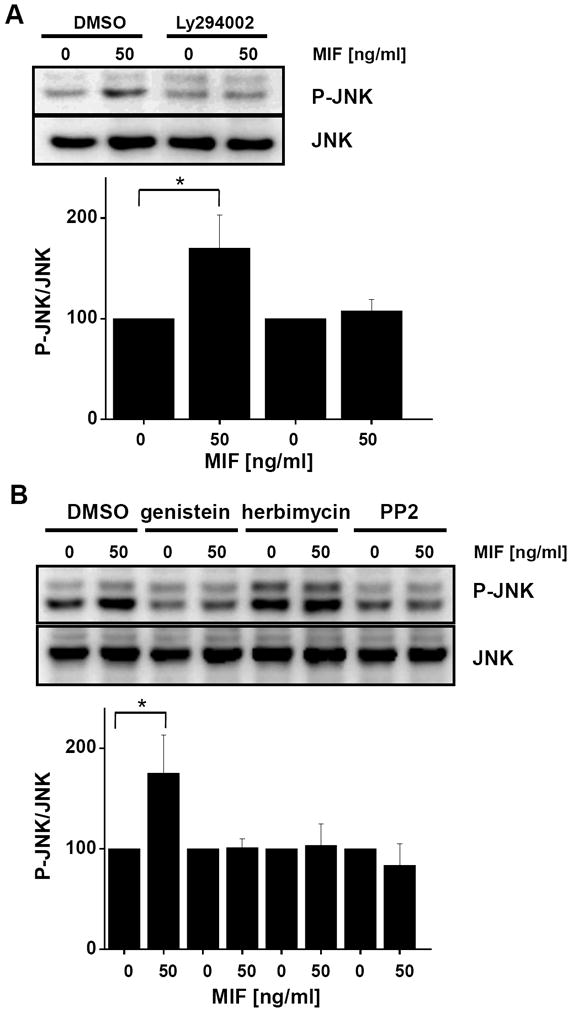
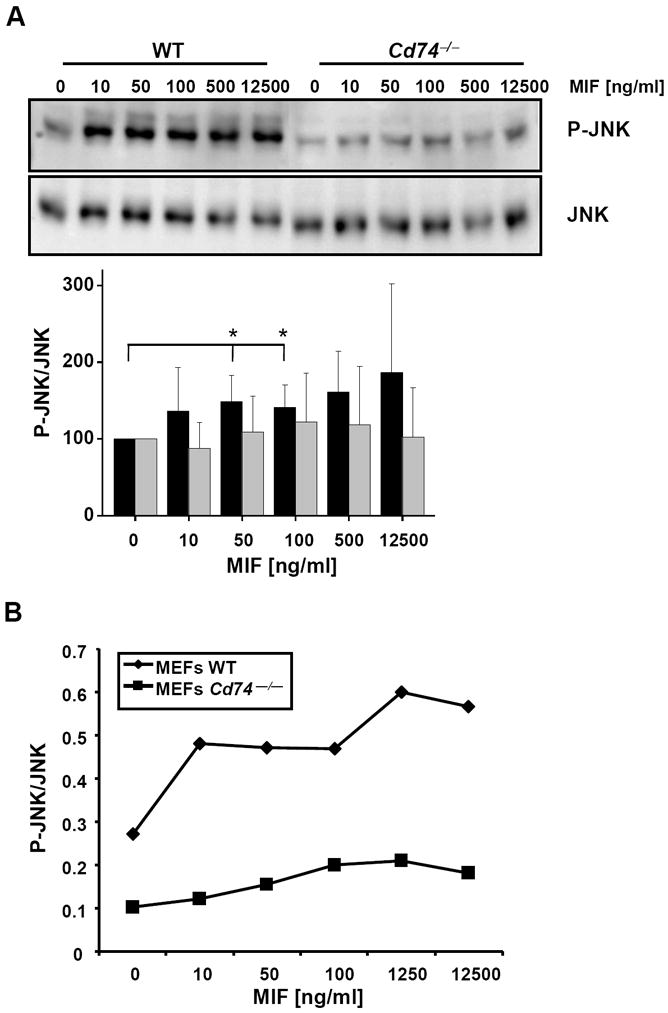
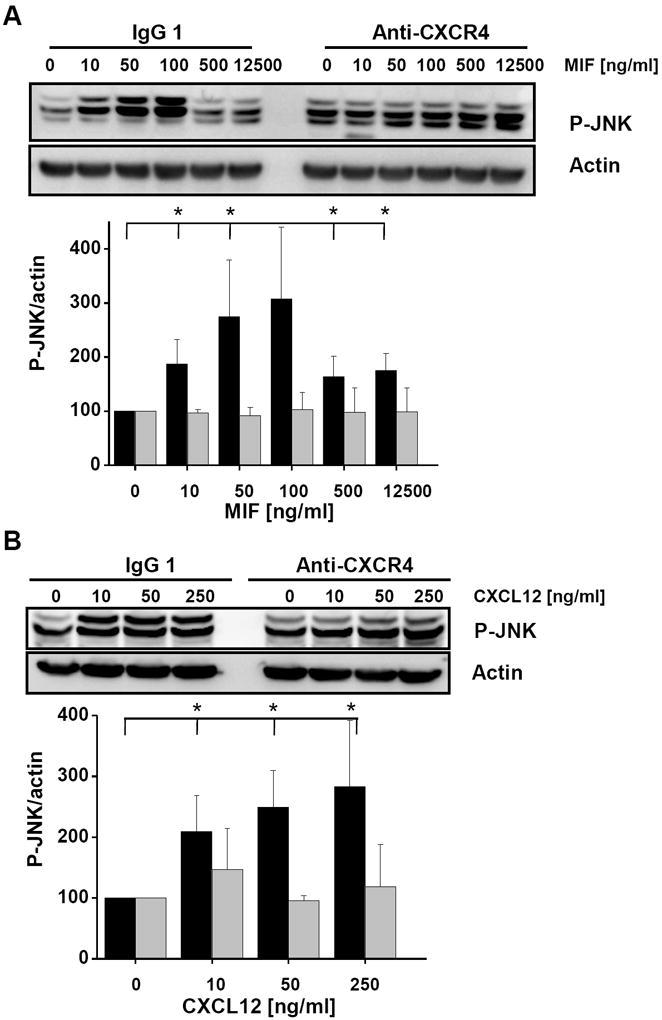
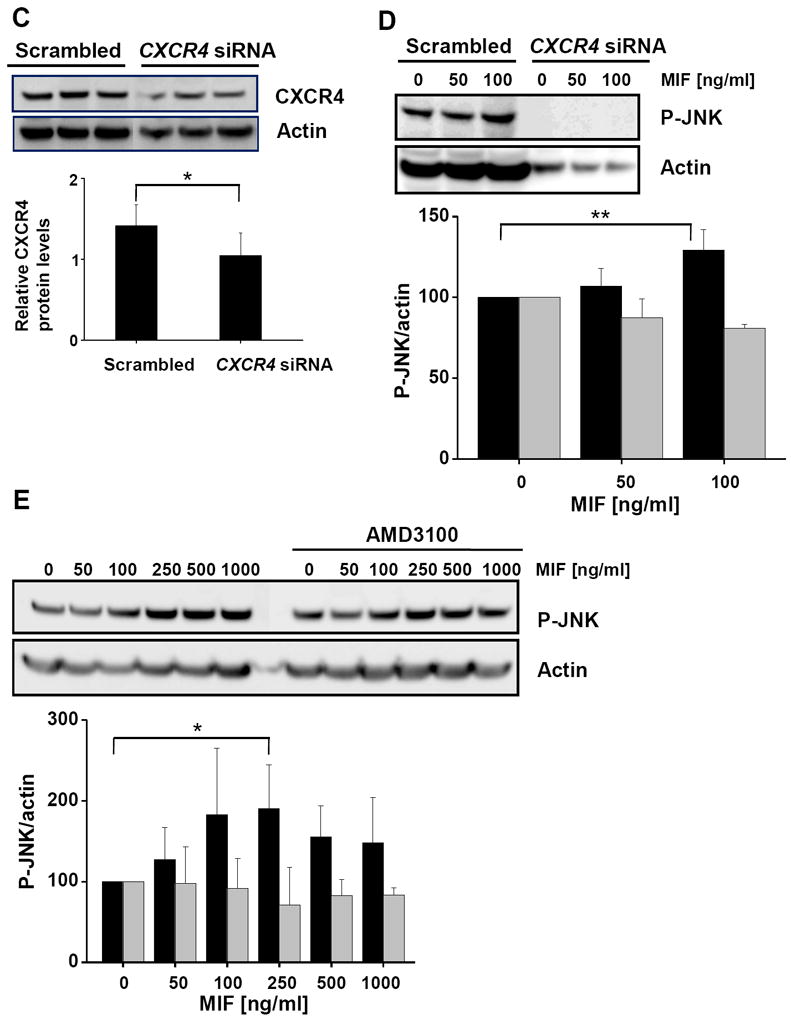
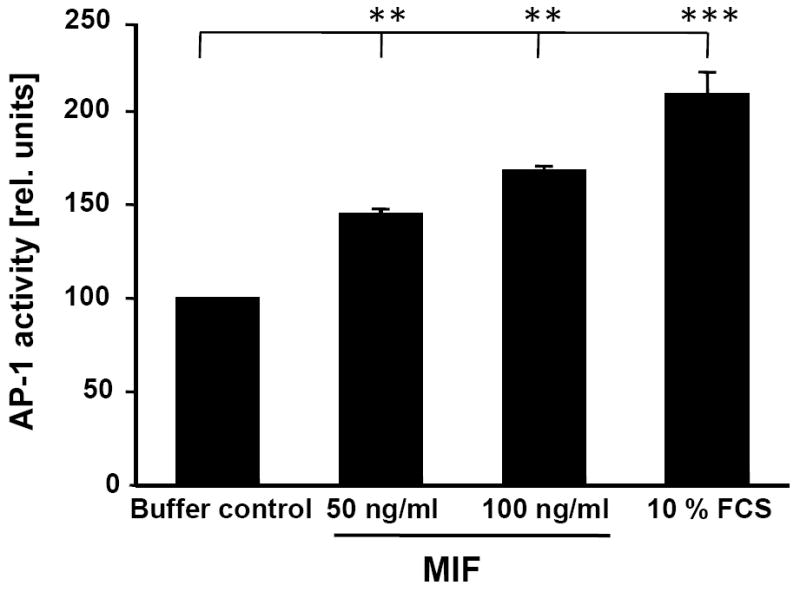
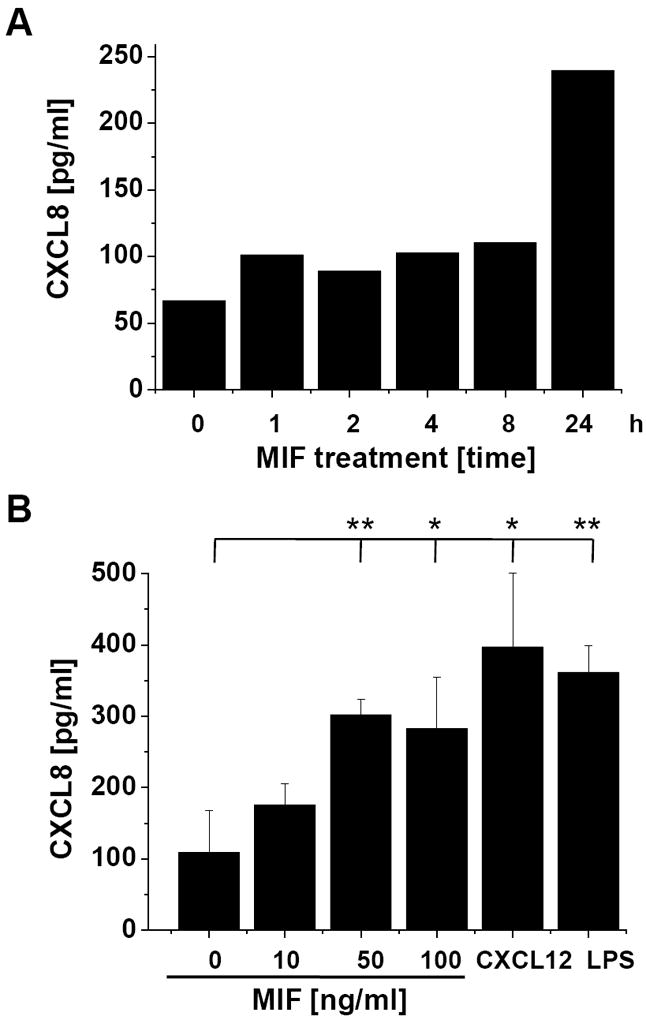
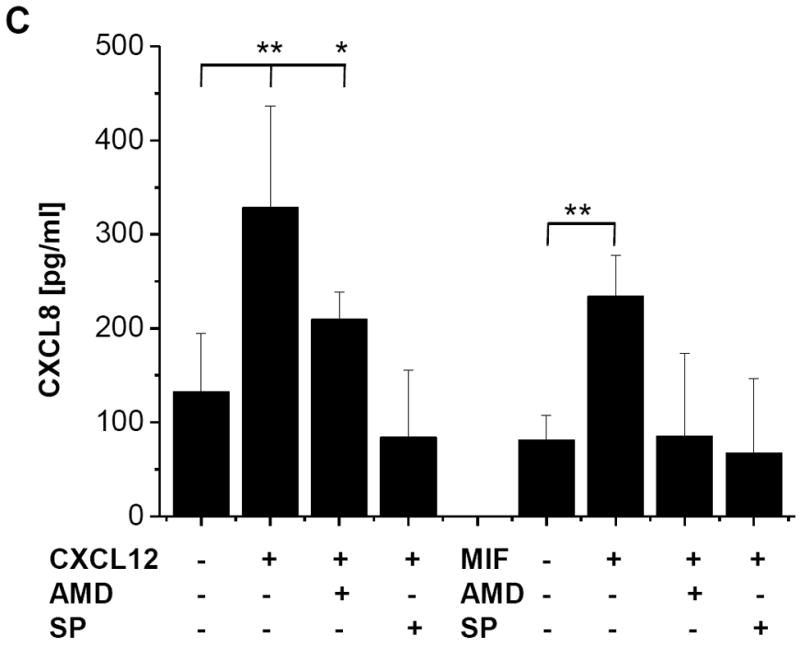
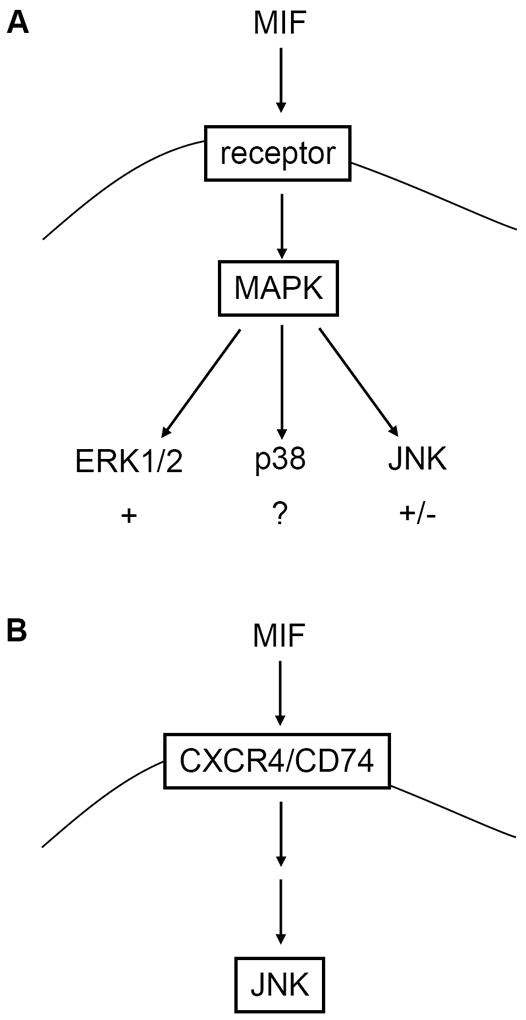
c-Jun N-terminal kinase (JNK) is a member of the mitogen-activated protein kinase (MAPK) family and controls essential processes such as inflammation, cell differentiation, and apoptosis. JNK signalling is triggered by extracellular signals such as cytokines and environmental stresses. Macrophage migration inhibitory factor (MIF) is a pleiotropic pro-inflammatory cytokine with chemokine-like functions in leukocyte recruitment and atherosclerosis. MIF promotes MAPK signalling through ERK1/2, while it can either activate or inhibit JNK phosphorylation, depending on the cell type and underlying stimulation context. MIF activities are mediated by non-cognate interactions with the CXC chemokine receptors CXCR2 and CXCR4 or by ligation of CD74, which is the cell surface expressed form of the class II invariant chain. ERK1/2 signalling stimulated by MIF is dependent on CD74, but the receptor pathway involved in MIF activation of the JNK pathway is unknown. Here we comprehensively characterize the stimulatory effect of MIF on the canonical JNK/c-Jun/AP-1 pathway in fibroblasts and T cell lines and identify the upstream signalling components. Physiological concentrations of recombinant MIF triggered the phosphorylation of JNK and c-Jun and rapidly activated AP-1. In T cells, MIF-mediated activation of the JNK pathway led to upregulated gene expression of the inflammatory chemokine CXCL8. Activation of JNK signalling by MIF involved the upstream kinases PI3K and SRC and was found to be dependent on CXCR4 and CD74. Together, these data show that the CXCR4/CD74/SRC/PI3K axis mediates a rapid and transient activation of the JNK pathway as triggered by the inflammatory cytokine MIF in T cells and fibroblasts.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
LPS-mediated cell surface expression of CD74 promotes the proliferation of B cells in response to MIF.Cell Signal. 2018 Jun;46:32-42. doi: 10.1016/j.cellsig.2018.02.010. Epub 2018 Feb 21. Cell Signal. 2018. PMID: 29476963
-
A functional heteromeric MIF receptor formed by CD74 and CXCR4.FEBS Lett. 2009 Sep 3;583(17):2749-57. doi: 10.1016/j.febslet.2009.07.058. Epub 2009 Aug 6. FEBS Lett. 2009. PMID: 19665027 Free PMC article.
-
CD74 is a functional MIF receptor on activated CD4+ T cells.Cell Mol Life Sci. 2024 Jul 11;81(1):296. doi: 10.1007/s00018-024-05338-5. Cell Mol Life Sci. 2024. PMID: 38992165 Free PMC article.
-
MIF, CD74 and other partners in kidney disease: tales of a promiscuous couple.Cytokine Growth Factor Rev. 2013 Feb;24(1):23-40. doi: 10.1016/j.cytogfr.2012.08.001. Epub 2012 Sep 7. Cytokine Growth Factor Rev. 2013. PMID: 22959722 Review.
-
MIF and CD74 - suitability as clinical biomarkers.Mini Rev Med Chem. 2014;14(14):1125-31. doi: 10.2174/1389557515666150203143317. Mini Rev Med Chem. 2014. PMID: 25643609 Review.
Cited by
-
Macrophage migration inhibitory factor produced by the tumour stroma but not by tumour cells regulates angiogenesis in the B16-F10 melanoma model.Br J Cancer. 2012 Oct 23;107(9):1498-505. doi: 10.1038/bjc.2012.392. Epub 2012 Sep 6. Br J Cancer. 2012. PMID: 22955855 Free PMC article.
-
Role of macrophage migration inhibitory factor (MIF) in pollen-induced allergic conjunctivitis and pollen dermatitis in mice.PLoS One. 2015 Feb 3;10(2):e0115593. doi: 10.1371/journal.pone.0115593. eCollection 2015. PLoS One. 2015. PMID: 25647395 Free PMC article.
-
Beta cell function: the role of macrophage migration inhibitory factor.Immunol Res. 2012 Apr;52(1-2):81-8. doi: 10.1007/s12026-012-8281-y. Immunol Res. 2012. PMID: 22388641 Review.
-
Macrophage migration inhibitory factor promotes eosinophil accumulation and tissue remodeling in eosinophilic esophagitis.Mucosal Immunol. 2015 Sep;8(5):1154-65. doi: 10.1038/mi.2015.6. Epub 2015 Feb 25. Mucosal Immunol. 2015. PMID: 25712805 Free PMC article. Clinical Trial.
-
Apis mellifera anatoliaca Venom Exerted Anti-Inflammatory Activity on LPS-Stimulated Mammalian Macrophages by Reducing the Production of the Inflammatory Cytokines.Appl Biochem Biotechnol. 2023 May;195(5):3194-3205. doi: 10.1007/s12010-022-04284-x. Epub 2022 Dec 27. Appl Biochem Biotechnol. 2023. PMID: 36574137
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
