

Phenotypic variability and genetic susceptibility to genomic disorders
- PMID: 20807775
- PMCID: PMC2953748
- DOI: 10.1093/hmg/ddq366
Phenotypic variability and genetic susceptibility to genomic disorders
Abstract
The duplication architecture of the human genome predisposes our species to recurrent copy number variation and disease. Emerging data suggest that this mechanism of mutation contributes to both common and rare diseases. Two features regarding this form of mutation have emerged. First, common structural polymorphisms create susceptible and protective chromosomal architectures. These structural polymorphisms occur at varying frequencies in populations, leading to different susceptibility and ethnic predilection. Second, a subset of rearrangements shows extreme variability in expressivity. We propose that two types of genomic disorders may be distinguished: syndromic forms where the phenotypic features are largely invariant and those where the same molecular lesion associates with a diverse set of diagnoses including epilepsy, schizophrenia, autism, intellectual disability and congenital malformations. Copy number variation analyses of patient genomes reveal that disease type and severity may be explained by the occurrence of additional rare events and their inheritance within families. We propose that the overall burden of copy number variants creates differing sensitized backgrounds during development leading to different thresholds and disease outcomes. We suggest that the accumulation of multiple high-penetrant alleles of low frequency may serve as a more general model for complex genetic diseases, posing a significant challenge for diagnostics and disease management.
Figures
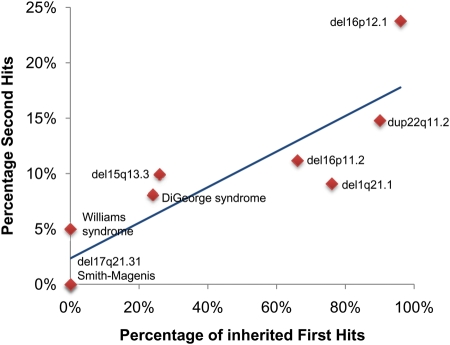

References
-
- Raeymaekers P., Timmerman V., Nelis E., De Jonghe P., Hoogendijk J.E., Baas F., Barker D.F., Martin J.J., De Visser M., Bolhuis P.A., et al. Duplication in chromosome 17p11.2 in Charcot–Marie–Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group . Neuromuscul. Disord. 1991;1:93–97. doi:10.1016/0960-8966(91)90055-W. - DOI - PubMed
-
- Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A., et al. DNA duplication associated with Charcot–Marie–Tooth disease type 1A. Cell. 1991;66:219–232. doi:10.1016/0092-8674(91)90613-4. - DOI - PubMed
-
- Chance P.F., Alderson M.K., Leppig K.A., Lensch M.W., Matsunami N., Smith B., Swanson P.D., Odelberg S.J., Disteche C.M., Bird T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi:10.1016/0092-8674(93)90058-X. - DOI - PubMed
-
- Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi:10.1016/S0168-9525(98)01555-8. - DOI - PubMed
-
- Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi:10.1126/science.1072047. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
