

A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons
- PMID: 20807776
- PMCID: PMC3298848
- DOI: 10.1093/hmg/ddq361
A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons
Abstract
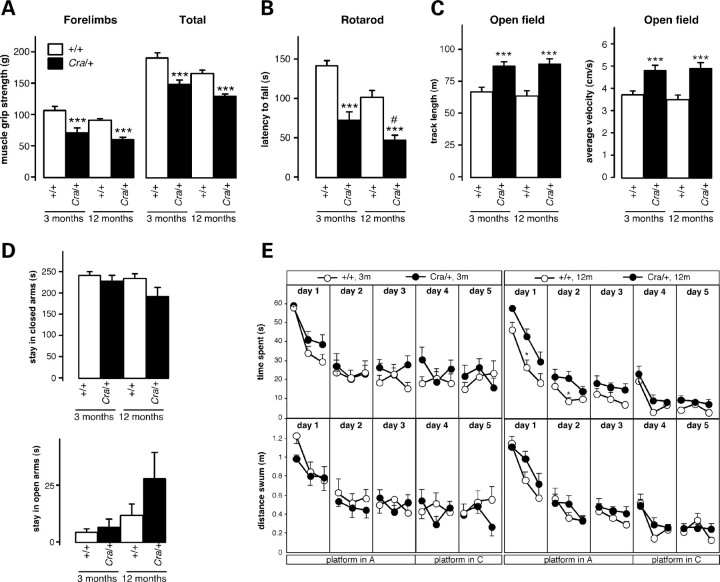
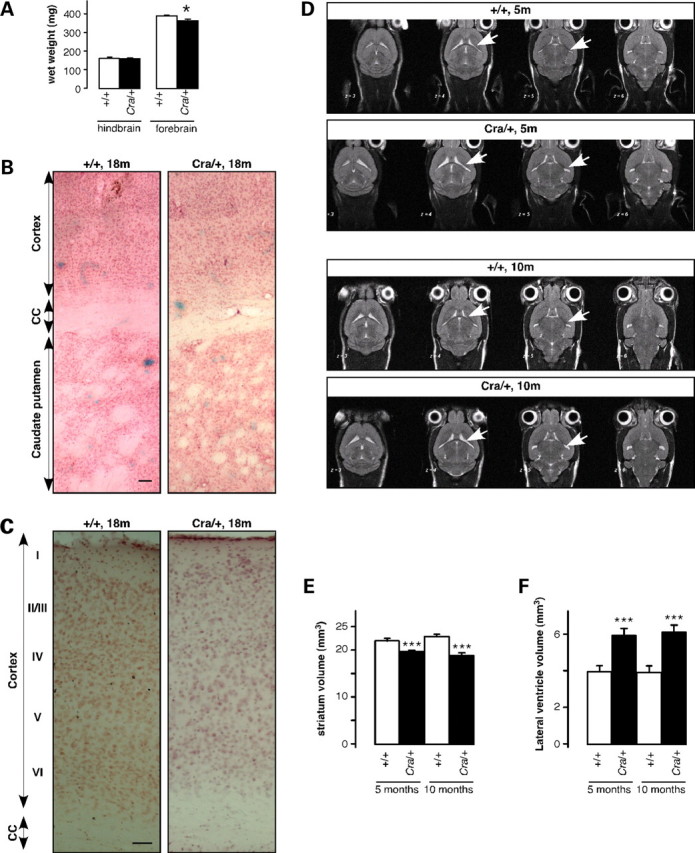
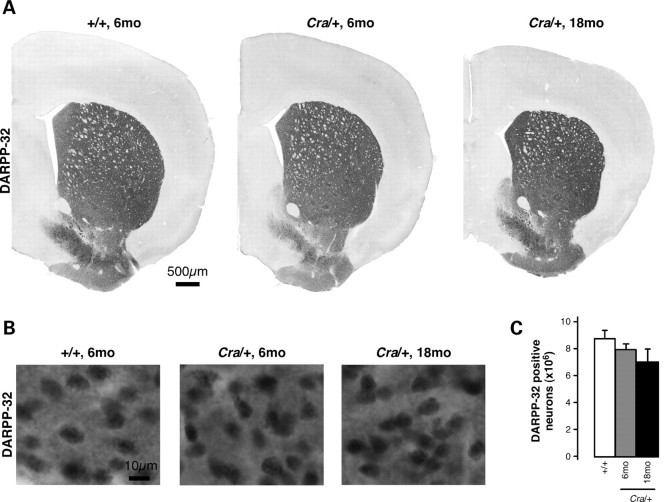
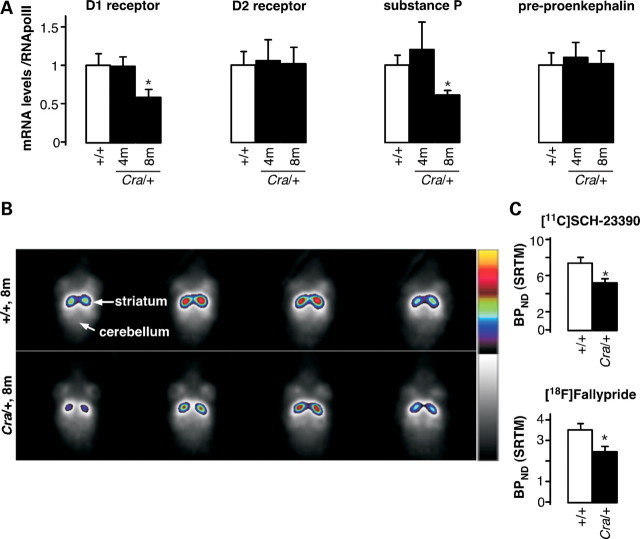
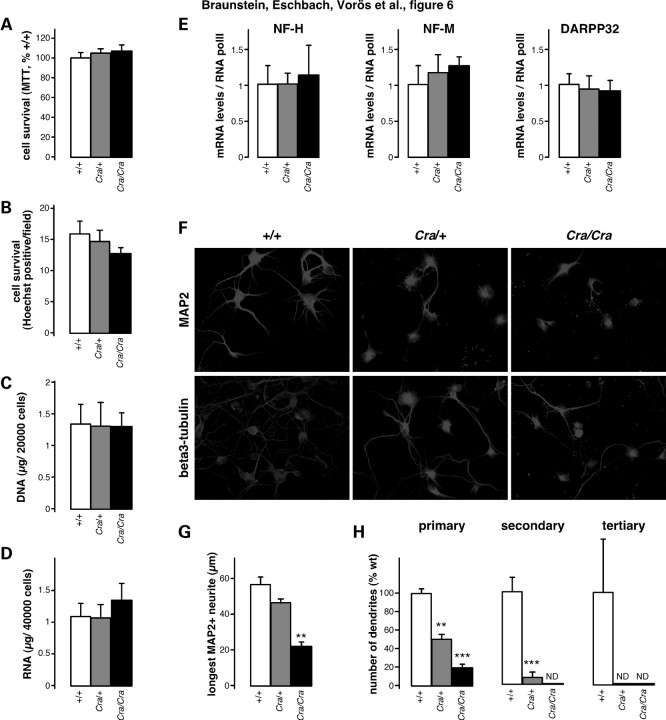
The molecular motor dynein and its associated regulatory subunit dynactin have been implicated in several neurodegenerative conditions of the basal ganglia, such as Huntington's disease (HD) and Perry syndrome, an atypical Parkinson-like disease. This pathogenic role has been largely postulated from the existence of mutations in the dynactin subunit p150(Glued). However, dynactin is also able to act independently of dynein, and there is currently no direct evidence linking dynein to basal ganglia degeneration. To provide such evidence, we used here a mouse strain carrying a point mutation in the dynein heavy chain gene that impairs retrograde axonal transport. These mice exhibited motor and behavioural abnormalities including hindlimb clasping, early muscle weakness, incoordination and hyperactivity. In vivo brain imaging using magnetic resonance imaging showed striatal atrophy and lateral ventricle enlargement. In the striatum, altered dopamine signalling, decreased dopamine D1 and D2 receptor binding in positron emission tomography SCAN and prominent astrocytosis were observed, although there was no neuronal loss either in the striatum or substantia nigra. In vitro, dynein mutant striatal neurons displayed strongly impaired neuritic morphology. Altogether, these findings provide a direct genetic evidence for the requirement of dynein for the morphology and function of striatal neurons. Our study supports a role for dynein dysfunction in the pathogenesis of neurodegenerative disorders of the basal ganglia, such as Perry syndrome and HD.
Figures

Similar articles
-
Low dietary protein content alleviates motor symptoms in mice with mutant dynactin/dynein-mediated neurodegeneration.Hum Mol Genet. 2015 Apr 15;24(8):2228-40. doi: 10.1093/hmg/ddu741. Epub 2014 Dec 30. Hum Mol Genet. 2015. PMID: 25552654 Free PMC article.
-
Autoradiographic study of striatal dopamine re-uptake sites and dopamine D1 and D2 receptors in a 6-hydroxydopamine and quinolinic acid double-lesion rat model of striatonigral degeneration (multiple system atrophy) and effects of embryonic ventral mesencephalic, striatal or co-grafts.Neuroscience. 2000;95(2):377-88. doi: 10.1016/s0306-4522(99)00457-1. Neuroscience. 2000. PMID: 10658617
-
Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington's disease.J Neurochem. 2005 Jun;93(5):1057-68. doi: 10.1111/j.1471-4159.2005.03047.x. J Neurochem. 2005. PMID: 15934928
-
Cytoplasmic dynein/dynactin function and dysfunction in motor neurons.Int J Dev Neurosci. 2006 Apr-May;24(2-3):103-11. doi: 10.1016/j.ijdevneu.2005.11.013. Epub 2006 Jan 6. Int J Dev Neurosci. 2006. PMID: 16406469 Review.
-
Striatal and extrastriatal dopamine in the basal ganglia: an overview of its anatomical organization in normal and Parkinsonian brains.Mov Disord. 2008;23 Suppl 3:S534-47. doi: 10.1002/mds.22027. Mov Disord. 2008. PMID: 18781680 Review.
Cited by
-
PATHOLOGIES OF AXONAL TRANSPORT IN NEURODEGENERATIVE DISEASES.Transl Neurosci. 2012 Dec 1;3(4):355-372. doi: 10.2478/s13380-012-0044-7. Transl Neurosci. 2012. PMID: 23750323 Free PMC article.
-
Low dietary protein content alleviates motor symptoms in mice with mutant dynactin/dynein-mediated neurodegeneration.Hum Mol Genet. 2015 Apr 15;24(8):2228-40. doi: 10.1093/hmg/ddu741. Epub 2014 Dec 30. Hum Mol Genet. 2015. PMID: 25552654 Free PMC article.
-
Dynein mutations associated with hereditary motor neuropathies impair mitochondrial morphology and function with age.Neurobiol Dis. 2013 Oct;58:220-30. doi: 10.1016/j.nbd.2013.05.015. Epub 2013 Jun 4. Neurobiol Dis. 2013. PMID: 23742762 Free PMC article.
-
Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites.J Neurosci. 2013 Apr 10;33(15):6298-309. doi: 10.1523/JNEUROSCI.2033-12.2013. J Neurosci. 2013. PMID: 23575829 Free PMC article.
-
Shank3 Transgenic and Prenatal Zinc-Deficient Autism Mouse Models Show Convergent and Individual Alterations of Brain Structures in MRI.Front Neural Circuits. 2019 Feb 22;13:6. doi: 10.3389/fncir.2019.00006. eCollection 2019. Front Neural Circuits. 2019. PMID: 30853900 Free PMC article.
References
-
- Morfini G.A., Burns M., Binder L.I., Kanaan N.M., LaPointe N., Bosco D.A., Brown R.H., Jr, Brown H., Tiwari A., Hayward L., et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009;29:12776–12786. doi:10.1523/JNEUROSCI.3463-09.2009. - DOI - PMC - PubMed
-
- Chevalier-Larsen E., Holzbaur E.L. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta. 2006;1762:1094–1108. - PubMed
-
- Levy J.R., Holzbaur E.L. Cytoplasmic dynein/dynactin function and dysfunction in motor neurons. Int. J. Dev. Neurosci. 2006;24:103–111. doi:10.1016/j.ijdevneu.2005.11.013. - DOI - PubMed
-
- LaMonte B.H., Wallace K.E., Holloway B.A., Shelly S.S., Ascano J., Tokito M., Van Winkle T., Howland D.S., Holzbaur E.L. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi:10.1016/S0896-6273(02)00696-7. - DOI - PubMed
-
- Teuling E., van Dis V., Wulf P.S., Haasdijk E.D., Akhmanova A., Hoogenraad C.C., Jaarsma D. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum. Mol. Genet. 2008;17:2849–2862. doi:10.1093/hmg/ddn182. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
