

Nonlinear modelling of cancer: bridging the gap between cells and tumours
- PMID: 20808719
- PMCID: PMC2929802
- DOI: 10.1088/0951-7715/23/1/r01
Nonlinear modelling of cancer: bridging the gap between cells and tumours
Abstract
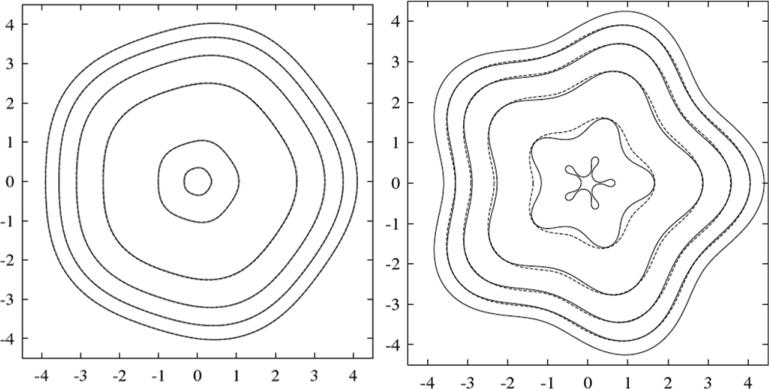
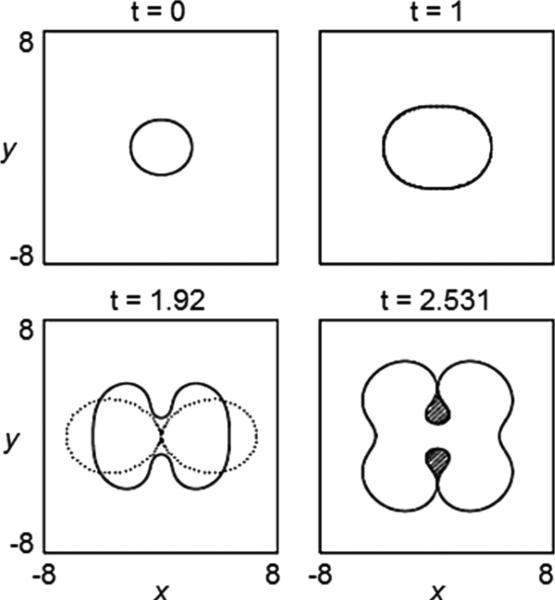
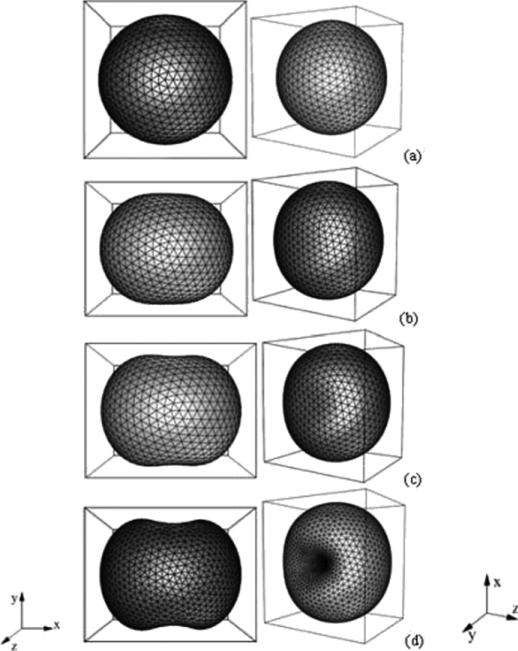
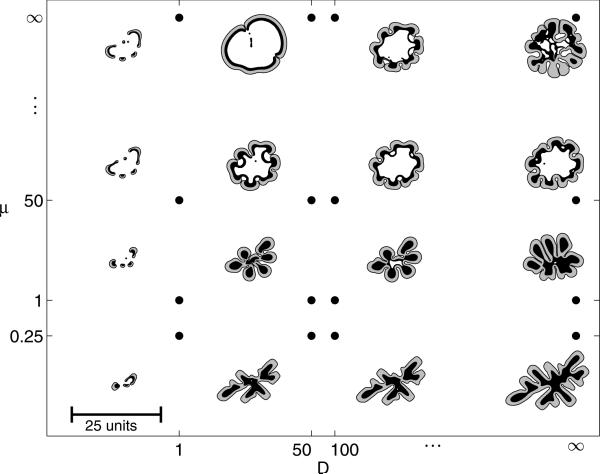
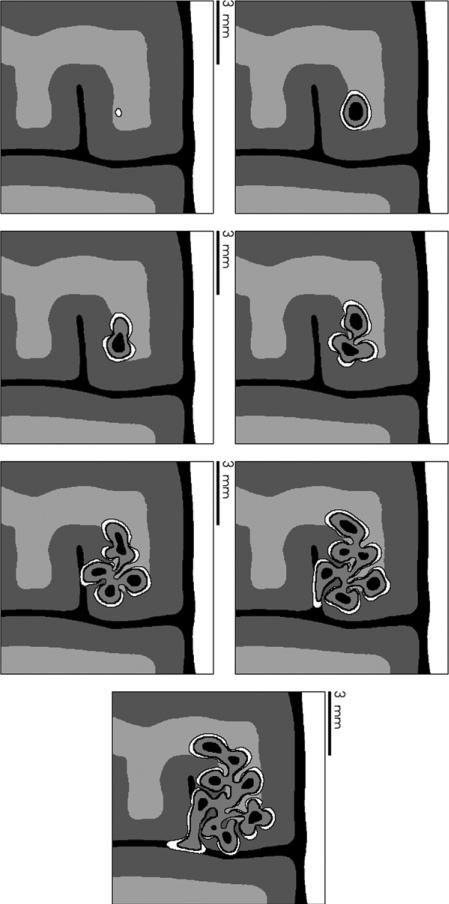
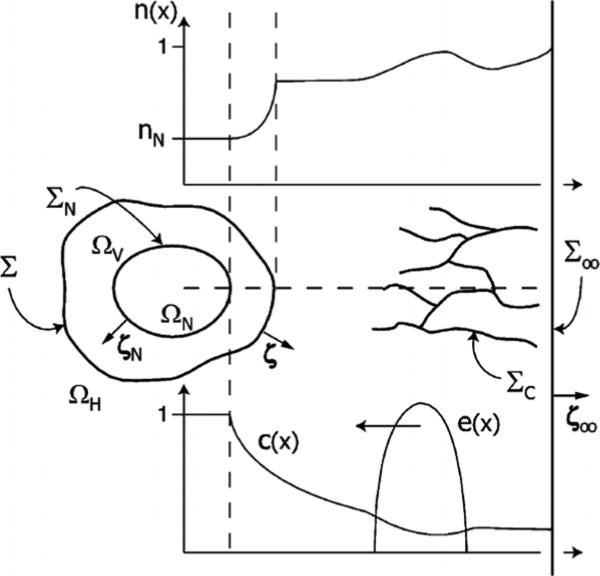
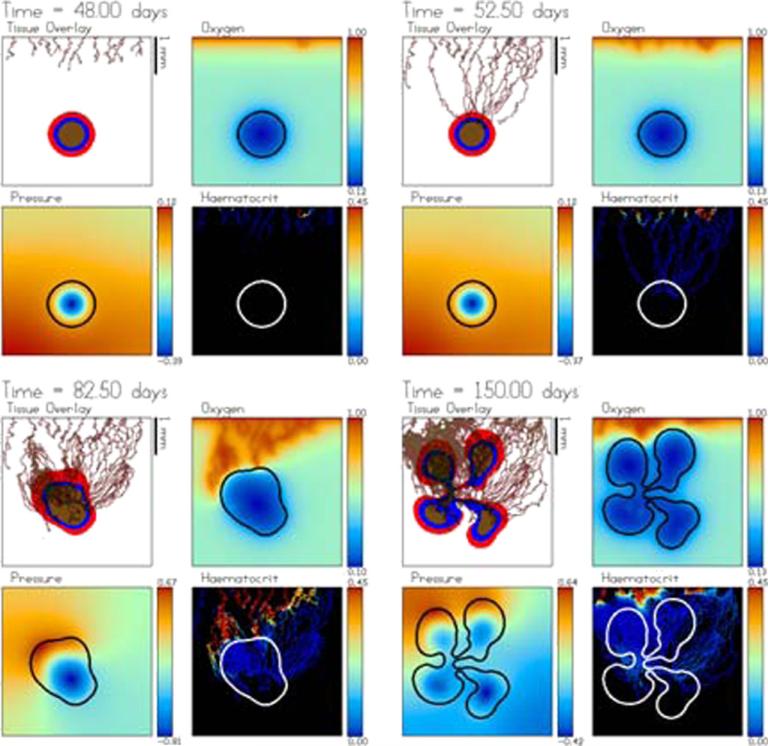
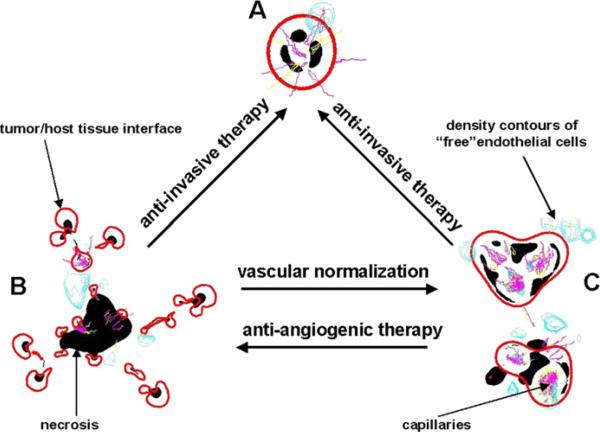
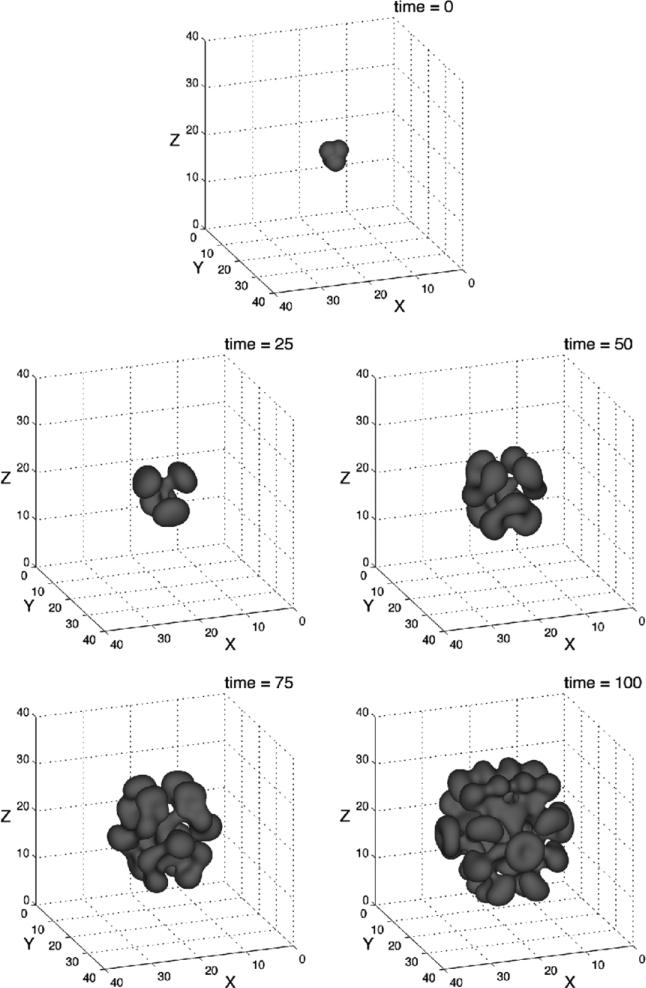
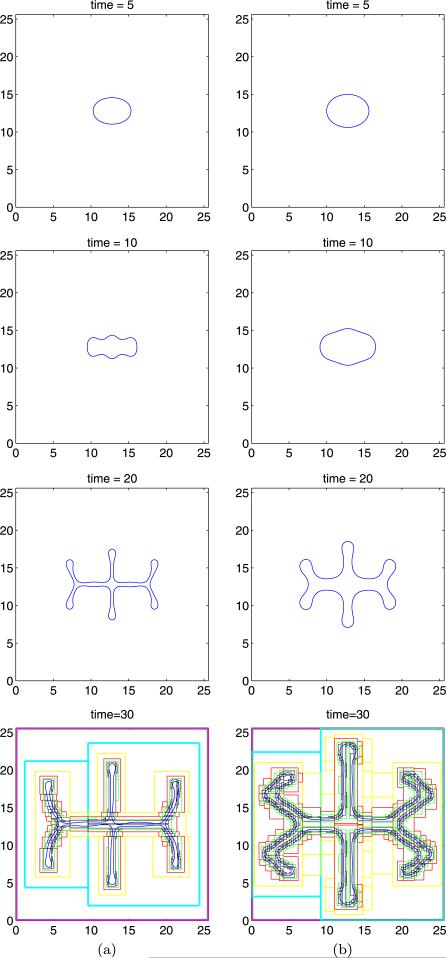
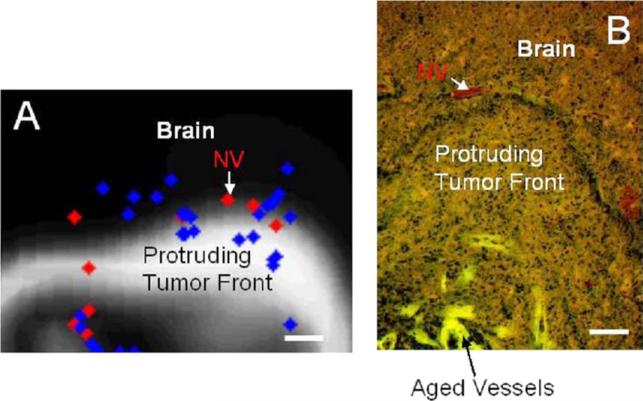
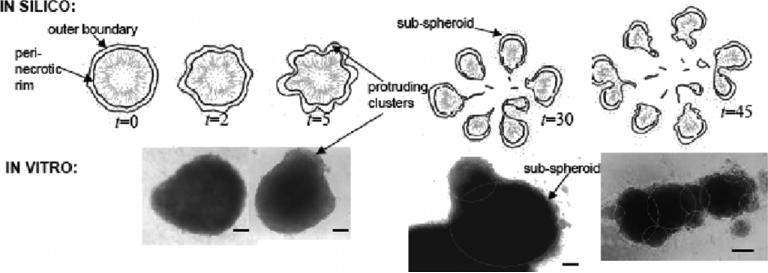
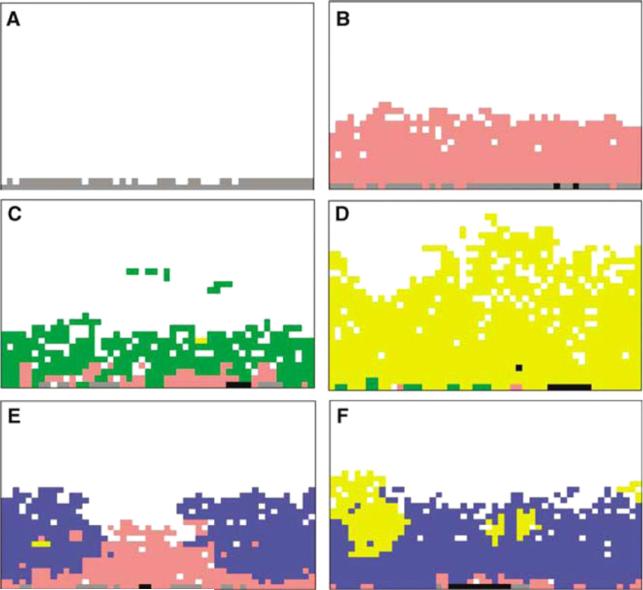
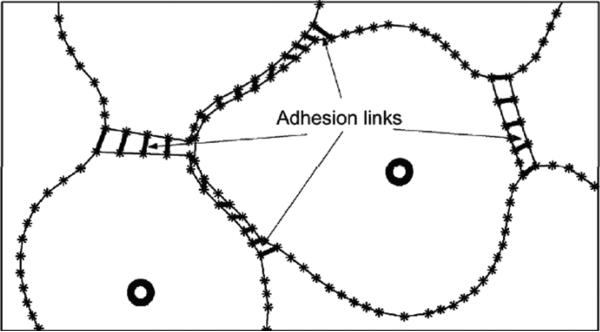
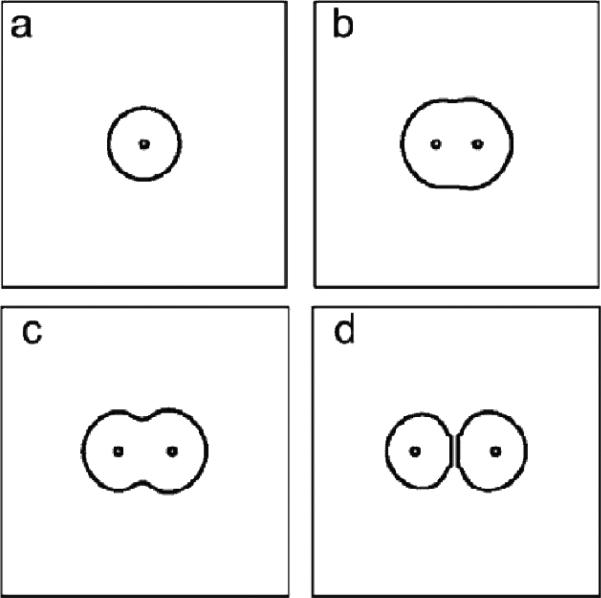
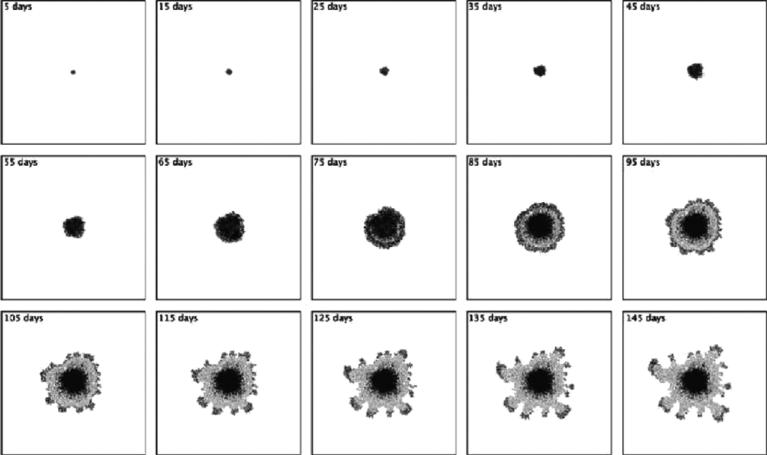
Despite major scientific, medical and technological advances over the last few decades, a cure for cancer remains elusive. The disease initiation is complex, and including initiation and avascular growth, onset of hypoxia and acidosis due to accumulation of cells beyond normal physiological conditions, inducement of angiogenesis from the surrounding vasculature, tumour vascularization and further growth, and invasion of surrounding tissue and metastasis. Although the focus historically has been to study these events through experimental and clinical observations, mathematical modelling and simulation that enable analysis at multiple time and spatial scales have also complemented these efforts. Here, we provide an overview of this multiscale modelling focusing on the growth phase of tumours and bypassing the initial stage of tumourigenesis. While we briefly review discrete modelling, our focus is on the continuum approach. We limit the scope further by considering models of tumour progression that do not distinguish tumour cells by their age. We also do not consider immune system interactions nor do we describe models of therapy. We do discuss hybrid-modelling frameworks, where the tumour tissue is modelled using both discrete (cell-scale) and continuum (tumour-scale) elements, thus connecting the micrometre to the centimetre tumour scale. We review recent examples that incorporate experimental data into model parameters. We show that recent mathematical modelling predicts that transport limitations of cell nutrients, oxygen and growth factors may result in cell death that leads to morphological instability, providing a mechanism for invasion via tumour fingering and fragmentation. These conditions induce selection pressure for cell survivability, and may lead to additional genetic mutations. Mathematical modelling further shows that parameters that control the tumour mass shape also control its ability to invade. Thus, tumour morphology may serve as a predictor of invasiveness and treatment prognosis.
Figures



References
-
- Abbott RG, Forrest S, Pienta KJ. Simulating the hallmarks of cancer. Art. Life. 2006;12:617–34. - PubMed
-
- Abia LM, Angulo O, Lopez-Marcos JC. Age-structured population models and their numerical solution. Ecol. Modelling. 2005;188:112–36.
-
- Adam JA. A simplified mathematical model of tumor growth. Math. Biosci. 1986;81:229–44.
-
- Adam JA. A mathematical model of tumor growth: II. Effects of geometry and spatial nonuniformity on stability. Math. Biosci. 1987;86:183–211.
-
- Adam JA. A mathematical model of tumor growth: III. Comparison with experiment. Math. Biosci. 1987;86:213–27.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous