

Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution
- PMID: 20808788
- PMCID: PMC2925886
- DOI: 10.1371/journal.pone.0012339
Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution
Abstract
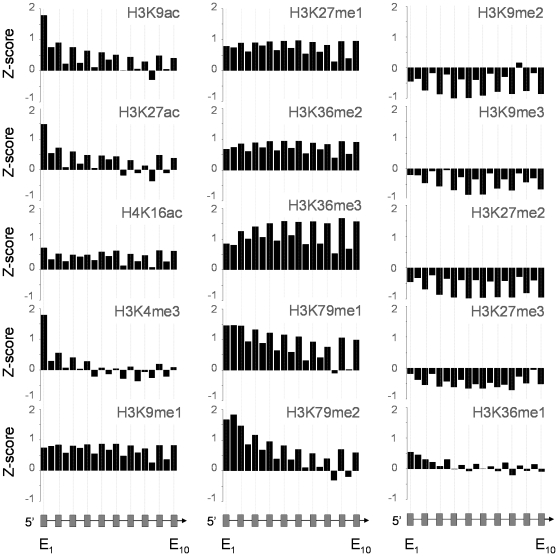
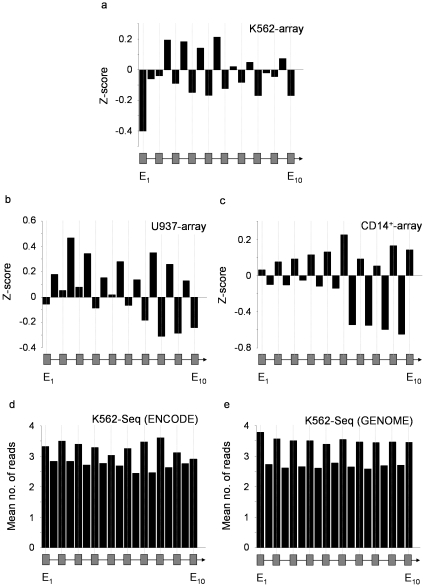
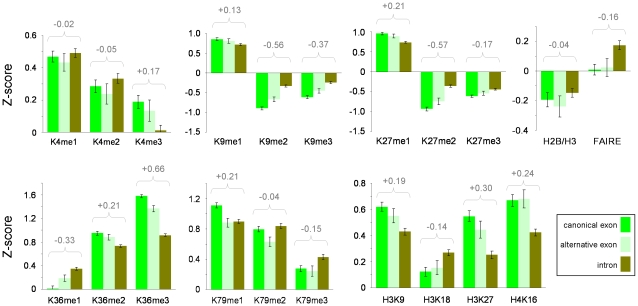
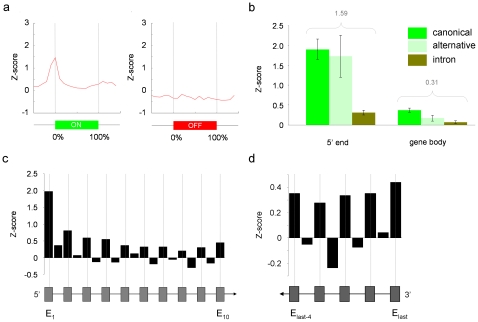
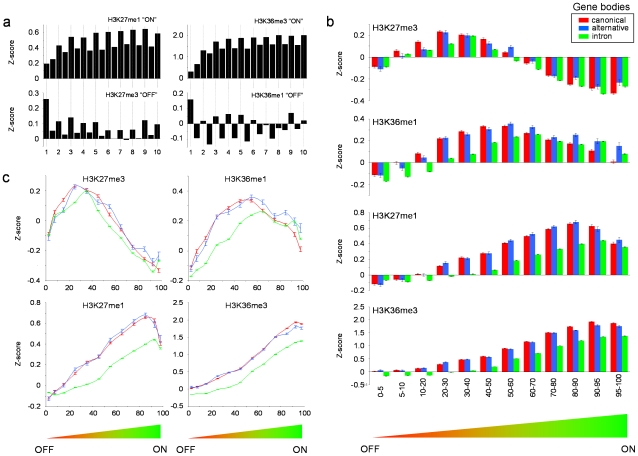
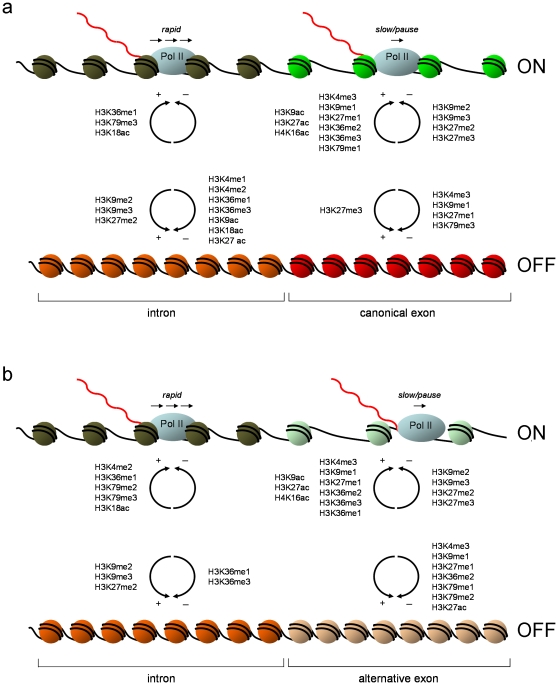
It has recently been shown that nucleosome distribution, histone modifications and RNA polymerase II (Pol II) occupancy show preferential association with exons ("exon-intron marking"), linking chromatin structure and function to co-transcriptional splicing in a variety of eukaryotes. Previous ChIP-sequencing studies suggested that these marking patterns reflect the nucleosomal landscape. By analyzing ChIP-chip datasets across the human genome in three cell types, we have found that this marking system is far more complex than previously observed. We show here that a range of histone modifications and Pol II are preferentially associated with exons. However, there is noticeable cell-type specificity in the degree of exon marking by histone modifications and, surprisingly, this is also reflected in some histone modifications patterns showing biases towards introns. Exon-intron marking is laid down in the absence of transcription on silent genes, with some marking biases changing or becoming reversed for genes expressed at different levels. Furthermore, the relationship of this marking system with splicing is not simple, with only some histone modifications reflecting exon usage/inclusion, while others mirror patterns of exon exclusion. By examining nucleosomal distributions in all three cell types, we demonstrate that these histone modification patterns cannot solely be accounted for by differences in nucleosome levels between exons and introns. In addition, because of inherent differences between ChIP-chip array and ChIP-sequencing approaches, these platforms report different nucleosome distribution patterns across the human genome. Our findings confound existing views and point to active cellular mechanisms which dynamically regulate histone modification levels and account for exon-intron marking. We believe that these histone modification patterns provide links between chromatin accessibility, Pol II movement and co-transcriptional splicing.
Conflict of interest statement
Figures
Similar articles
-
Differential patterns of intronic and exonic DNA regions with respect to RNA polymerase II occupancy, nucleosome density and H3K36me3 marking in fission yeast.Genome Biol. 2011 Aug 22;12(8):R82. doi: 10.1186/gb-2011-12-8-r82. Genome Biol. 2011. PMID: 21859475 Free PMC article.
-
Chromatin organization marks exon-intron structure.Nat Struct Mol Biol. 2009 Sep;16(9):990-5. doi: 10.1038/nsmb.1659. Nat Struct Mol Biol. 2009. PMID: 19684600
-
Nucleosome positioning as a determinant of exon recognition.Nat Struct Mol Biol. 2009 Sep;16(9):996-1001. doi: 10.1038/nsmb.1658. Nat Struct Mol Biol. 2009. PMID: 19684599
-
Histone exchange, chromatin structure and the regulation of transcription.Nat Rev Mol Cell Biol. 2015 Mar;16(3):178-89. doi: 10.1038/nrm3941. Epub 2015 Feb 4. Nat Rev Mol Cell Biol. 2015. PMID: 25650798 Review.
-
Pause locally, splice globally.Trends Cell Biol. 2011 Jun;21(6):328-35. doi: 10.1016/j.tcb.2011.03.002. Epub 2011 Apr 27. Trends Cell Biol. 2011. PMID: 21530266 Review.
Cited by
-
Distinct patterns of epigenetic marks and transcription factor binding sites across promoters of sense-intronic long noncoding RNAs.J Genet. 2015 Mar;94(1):17-25. doi: 10.1007/s12041-015-0484-2. J Genet. 2015. PMID: 25846873
-
Chromatin's thread to alternative splicing regulation.Chromosoma. 2013 Dec;122(6):465-74. doi: 10.1007/s00412-013-0425-x. Epub 2013 Aug 3. Chromosoma. 2013. PMID: 23912688 Review.
-
H2B ubiquitylation modulates spliceosome assembly and function in budding yeast.Biol Cell. 2014 Apr;106(4):126-38. doi: 10.1111/boc.201400003. Epub 2014 Feb 25. Biol Cell. 2014. PMID: 24476359 Free PMC article.
-
Co-transcriptional regulation of alternative pre-mRNA splicing.Biochim Biophys Acta. 2012 Jul;1819(7):673-83. doi: 10.1016/j.bbagrm.2012.01.014. Epub 2012 Feb 2. Biochim Biophys Acta. 2012. PMID: 22326677 Free PMC article. Review.
-
Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements.Cell Mol Life Sci. 2012 Nov;69(21):3613-34. doi: 10.1007/s00018-012-0990-9. Epub 2012 Apr 27. Cell Mol Life Sci. 2012. PMID: 22538991 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
