

Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing
- PMID: 20808830
- PMCID: PMC2924888
- DOI: 10.1371/journal.pone.0012303
Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing
Abstract
We used ultra-deep sequencing to obtain tens of thousands of HIV-1 sequences from regions targeted by CD8+ T lymphocytes from longitudinal samples from three acutely infected subjects, and modeled viral evolution during the critical first weeks of infection. Previous studies suggested that a single virus established productive infection, but these conclusions were tempered because of limited sampling; now, we have greatly increased our confidence in this observation through modeling the observed earliest sample diversity based on vastly more extensive sampling. Conventional sequencing of HIV-1 from acute/early infection has shown different patterns of escape at different epitopes; we investigated the earliest escapes in exquisite detail. Over 3-6 weeks, ultradeep sequencing revealed that the virus explored an extraordinary array of potential escape routes in the process of evading the earliest CD8 T-lymphocyte responses--using 454 sequencing, we identified over 50 variant forms of each targeted epitope during early immune escape, while only 2-7 variants were detected in the same samples via conventional sequencing. In contrast to the diversity seen within epitopes, non-epitope regions, including the Envelope V3 region, which was sequenced as a control in each subject, displayed very low levels of variation. In early infection, in the regions sequenced, the consensus forms did not have a fitness advantage large enough to trigger reversion to consensus amino acids in the absence of immune pressure. In one subject, a genetic bottleneck was observed, with extensive diversity at the second time point narrowing to two dominant escape forms by the third time point, all within two months of infection. Traces of immune escape were observed in the earliest samples, suggesting that immune pressure is present and effective earlier than previously reported; quantifying the loss rate of the founder virus suggests a direct role for CD8 T-lymphocyte responses in viral containment after peak viremia. Dramatic shifts in the frequencies of epitope variants during the first weeks of infection revealed a complex interplay between viral fitness and immune escape.
Conflict of interest statement
Figures
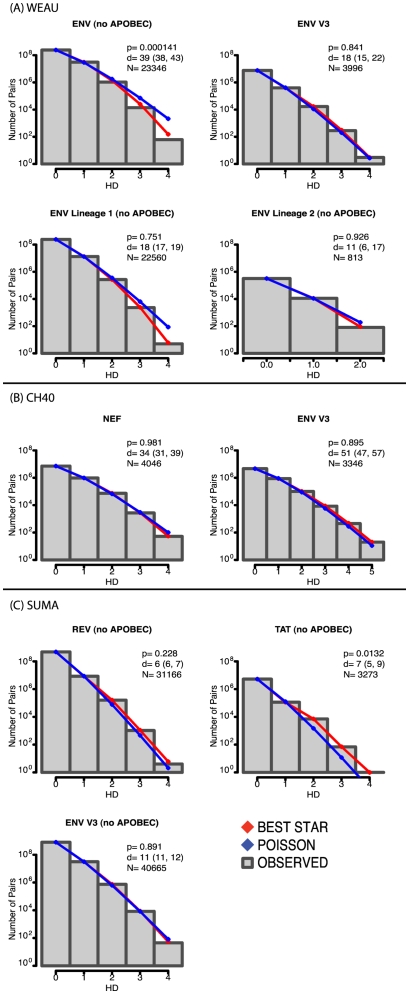
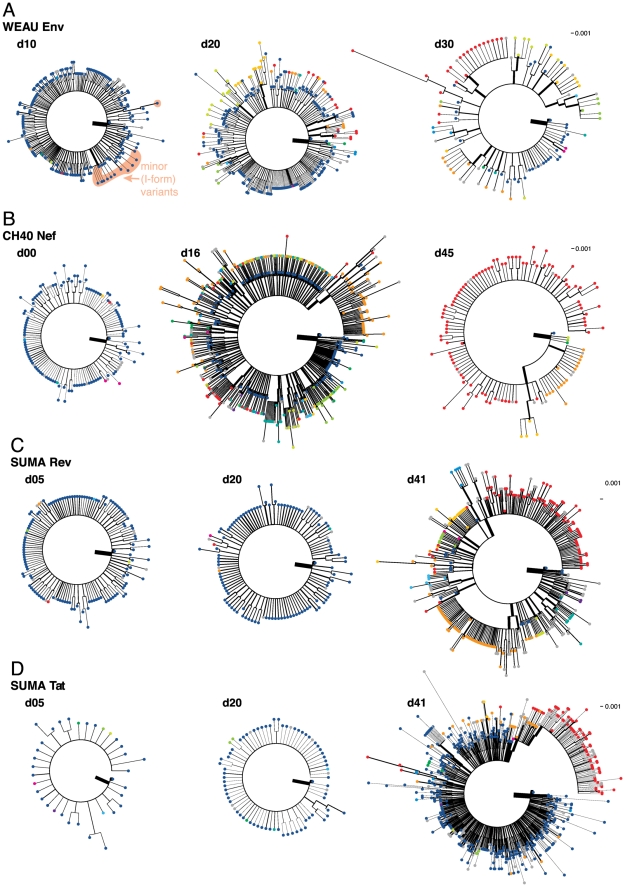
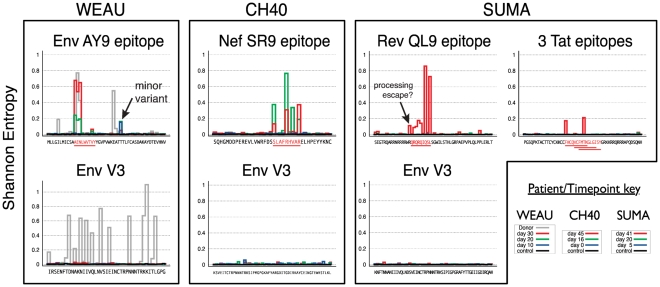
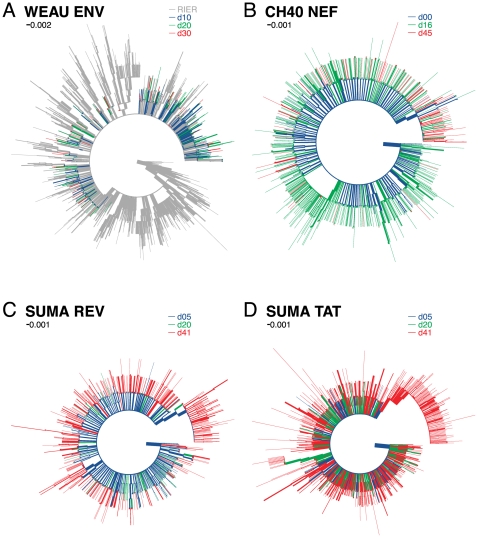
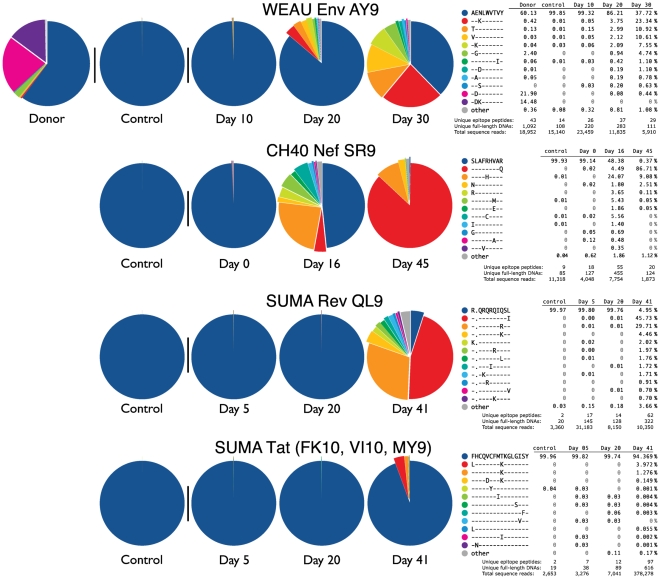
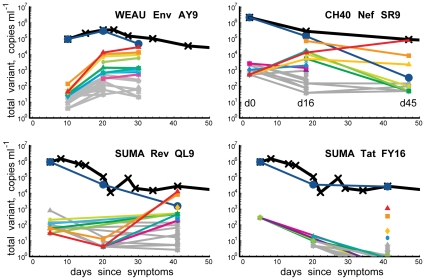
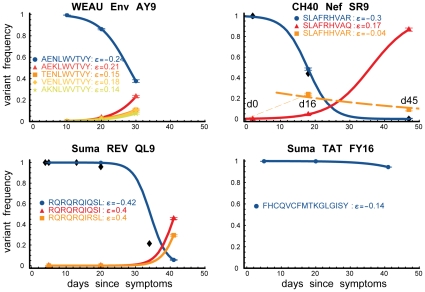
 , the rate of accumulation of variants. The dashed line in CH40 denotes accumulation of the SLAFHHVAR in the first 2 weeks of the infection (estimated minimal escape rate,
, the rate of accumulation of variants. The dashed line in CH40 denotes accumulation of the SLAFHHVAR in the first 2 weeks of the infection (estimated minimal escape rate,  = 0.44 day−1, doubling time 1.6 days). Our previous SGA-sequence-derived estimates of the founder sequence loss rate were lower, 0.13 day−1 (doubling time 5.3 days) and 0.10 day−1 (doubling time 6.9 days) for WEAU Env AY9 and SUMA TAT epitopes, respectively .
= 0.44 day−1, doubling time 1.6 days). Our previous SGA-sequence-derived estimates of the founder sequence loss rate were lower, 0.13 day−1 (doubling time 5.3 days) and 0.10 day−1 (doubling time 6.9 days) for WEAU Env AY9 and SUMA TAT epitopes, respectively .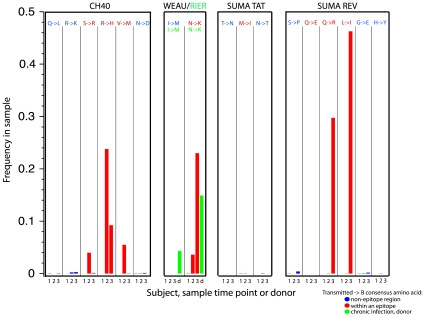
References
-
- Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–6. - PubMed
-
- Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
