

Disease-associated mutations that alter the RNA structural ensemble
- PMID: 20808897
- PMCID: PMC2924325
- DOI: 10.1371/journal.pgen.1001074
Disease-associated mutations that alter the RNA structural ensemble
Abstract
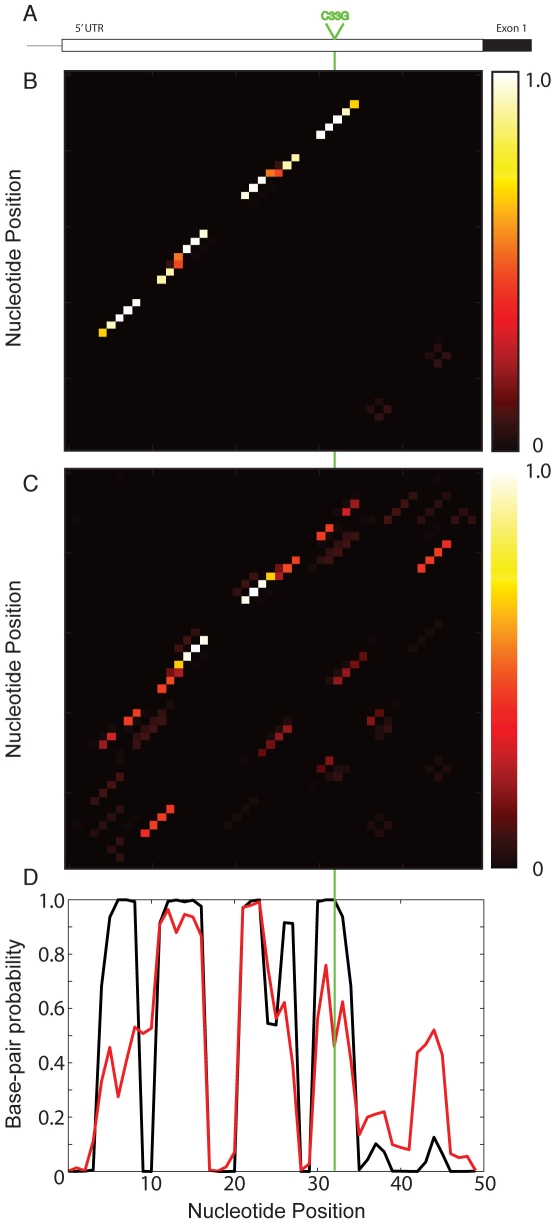
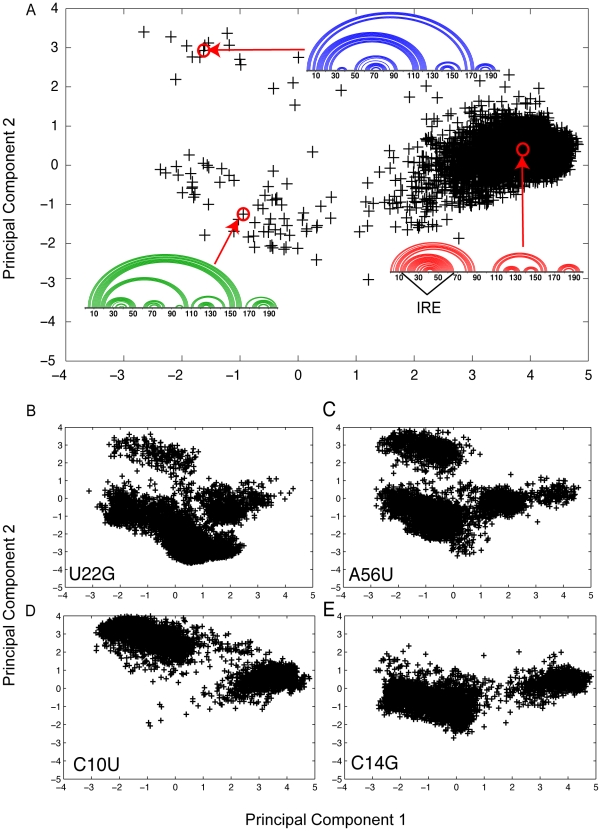
Genome-wide association studies (GWAS) often identify disease-associated mutations in intergenic and non-coding regions of the genome. Given the high percentage of the human genome that is transcribed, we postulate that for some observed associations the disease phenotype is caused by a structural rearrangement in a regulatory region of the RNA transcript. To identify such mutations, we have performed a genome-wide analysis of all known disease-associated Single Nucleotide Polymorphisms (SNPs) from the Human Gene Mutation Database (HGMD) that map to the untranslated regions (UTRs) of a gene. Rather than using minimum free energy approaches (e.g. mFold), we use a partition function calculation that takes into consideration the ensemble of possible RNA conformations for a given sequence. We identified in the human genome disease-associated SNPs that significantly alter the global conformation of the UTR to which they map. For six disease-states (Hyperferritinemia Cataract Syndrome, beta-Thalassemia, Cartilage-Hair Hypoplasia, Retinoblastoma, Chronic Obstructive Pulmonary Disease (COPD), and Hypertension), we identified multiple SNPs in UTRs that alter the mRNA structural ensemble of the associated genes. Using a Boltzmann sampling procedure for sub-optimal RNA structures, we are able to characterize and visualize the nature of the conformational changes induced by the disease-associated mutations in the structural ensemble. We observe in several cases (specifically the 5' UTRs of FTL and RB1) SNP-induced conformational changes analogous to those observed in bacterial regulatory Riboswitches when specific ligands bind. We propose that the UTR and SNP combinations we identify constitute a "RiboSNitch," that is a regulatory RNA in which a specific SNP has a structural consequence that results in a disease phenotype. Our SNPfold algorithm can help identify RiboSNitches by leveraging GWAS data and an analysis of the mRNA structural ensemble.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Comment in
-
Disease genetics: SNPs and the structural deficit.Nat Rev Genet. 2010 Oct;11(10):669. doi: 10.1038/nrg2871. Epub 2010 Sep 7. Nat Rev Genet. 2010. PMID: 20820181 No abstract available.
References
-
- Morton NE. Into the post-HapMap era. Adv Genet. 2008;60:727–742. - PubMed
-
- Mathew CG. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat Rev Genet. 2008;9:9–14. - PubMed
-
- Lee SH, van der Werf JH, Hayes BJ, Goddard ME, Visscher PM. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 2008;4:e1000231. doi: 10.1371/journal.pgen.1000231. - DOI - PMC - PubMed
-
- Lee ST, Choi KW, Yeo HT, Kim JW, Ki CS, et al. Identification of an Arg35X mutation in the PDCD10 gene in a patient with cerebral and multiple spinal cavernous malformations. J Neurol Sci. 2008;267:177–181. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
