

Extreme evolutionary disparities seen in positive selection across seven complex diseases
- PMID: 20808933
- PMCID: PMC2923198
- DOI: 10.1371/journal.pone.0012236
Extreme evolutionary disparities seen in positive selection across seven complex diseases
Abstract
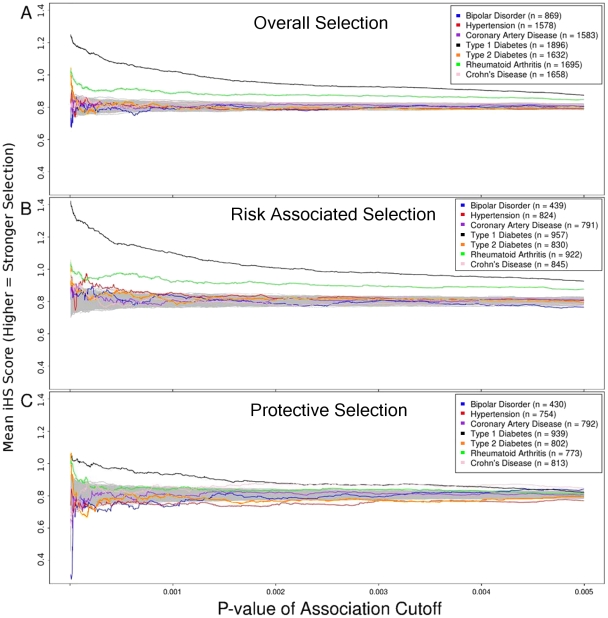
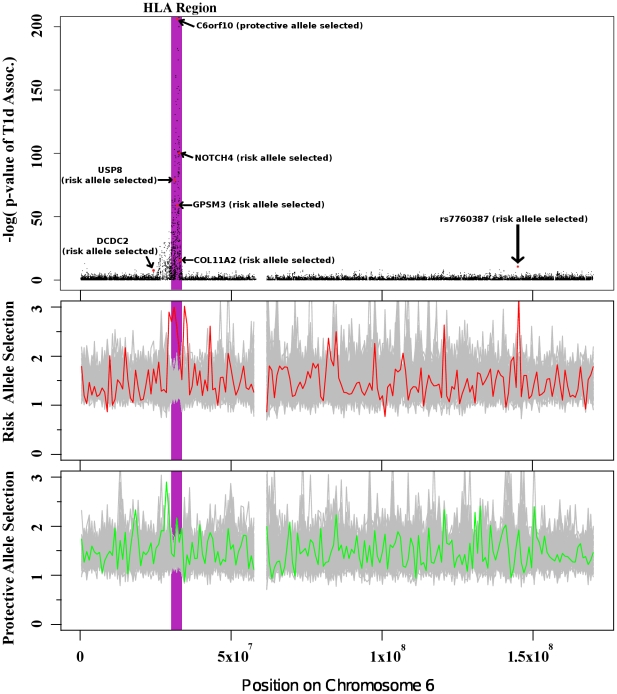
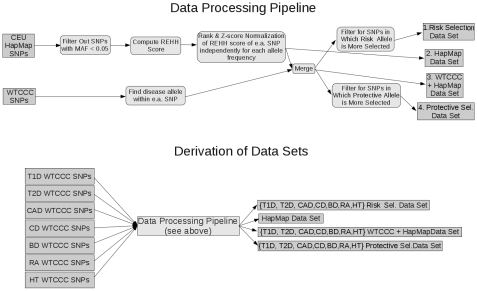
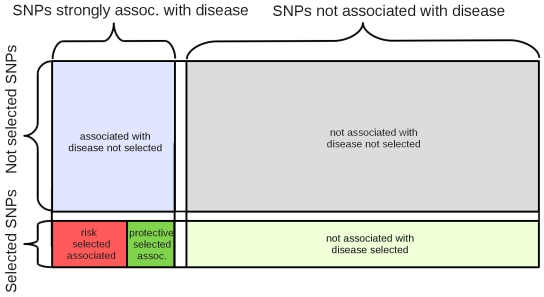
Positive selection is known to occur when the environment that an organism inhabits is suddenly altered, as is the case across recent human history. Genome-wide association studies (GWASs) have successfully illuminated disease-associated variation. However, whether human evolution is heading towards or away from disease susceptibility in general remains an open question. The genetic-basis of common complex disease may partially be caused by positive selection events, which simultaneously increased fitness and susceptibility to disease. We analyze seven diseases studied by the Wellcome Trust Case Control Consortium to compare evidence for selection at every locus associated with disease. We take a large set of the most strongly associated SNPs in each GWA study in order to capture more hidden associations at the cost of introducing false positives into our analysis. We then search for signs of positive selection in this inclusive set of SNPs. There are striking differences between the seven studied diseases. We find alleles increasing susceptibility to Type 1 Diabetes (T1D), Rheumatoid Arthritis (RA), and Crohn's Disease (CD) underwent recent positive selection. There is more selection in alleles increasing, rather than decreasing, susceptibility to T1D. In the 80 SNPs most associated with T1D (p-value <7.01 x 10(-5)) showing strong signs of positive selection, 58 alleles associated with disease susceptibility show signs of positive selection, while only 22 associated with disease protection show signs of positive selection. Alleles increasing susceptibility to RA are under selection as well. In contrast, selection in SNPs associated with CD favors protective alleles. These results inform the current understanding of disease etiology, shed light on potential benefits associated with the genetic-basis of disease, and aid in the efforts to identify causal genetic factors underlying complex disease.
Conflict of interest statement
Figures
References
-
- Maddox J. Migration out of Africa. Nature. 1994;372:32. - PubMed
-
- Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, et al. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. - PubMed
-
- Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet. 2009;10:872–878. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
