

DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia
- PMID: 20808941
- PMCID: PMC2922373
- DOI: 10.1371/journal.pone.0012197
DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia
Abstract
Background: Aberrant promoter DNA methylation has been shown to play a role in acute myeloid leukemia (AML) pathophysiology. However, further studies to discuss the prognostic value and the relationship of the epigenetic signatures with defined genomic rearrangements in acute myeloid leukemia are required.
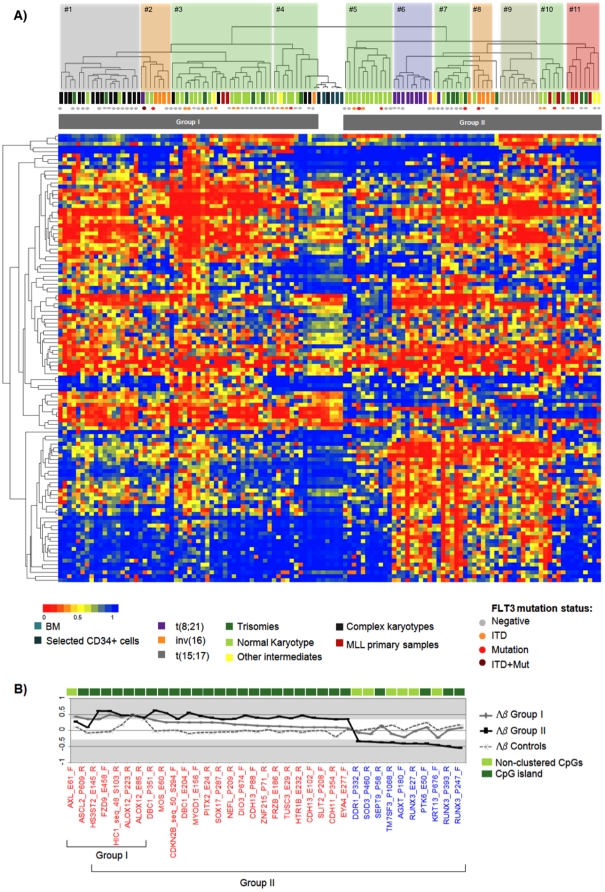
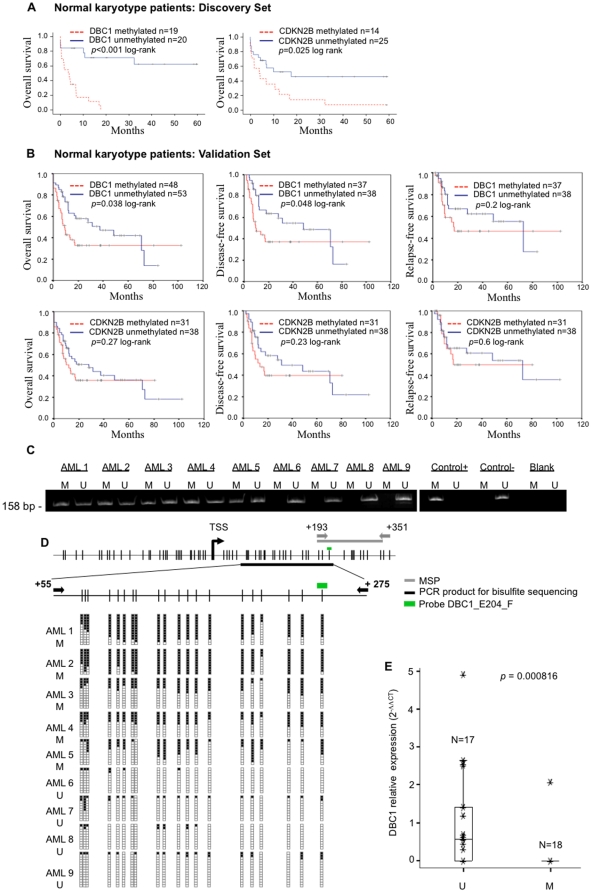
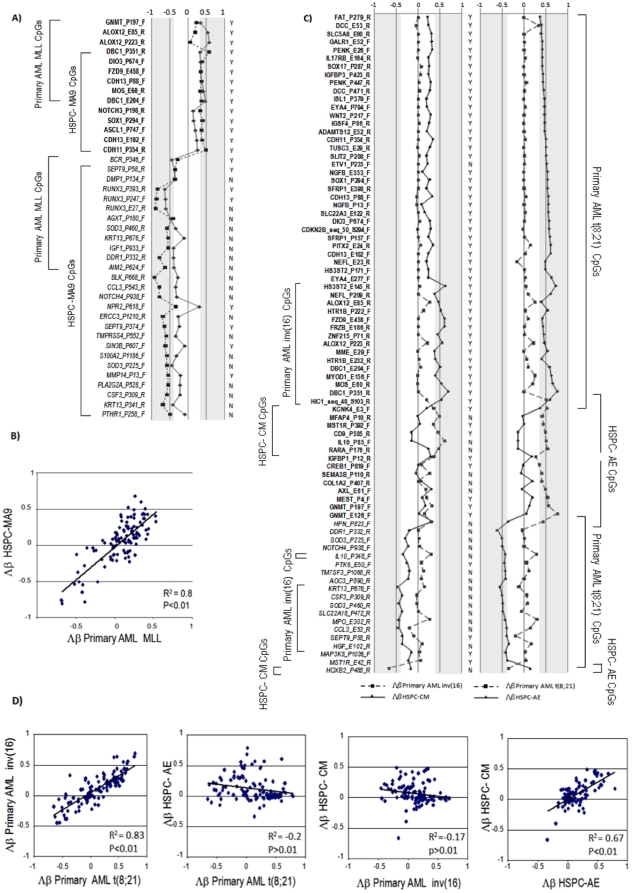
Methodology/principal findings: We carried out high-throughput methylation profiling on 116 de novo AML cases and we validated the significant biomarkers in an independent cohort of 244 AML cases. Methylation signatures were associated with the presence of a specific cytogenetic status. In normal karyotype cases, aberrant methylation of the promoter of DBC1 was validated as a predictor of the disease-free and overall survival. Furthermore, DBC1 expression was significantly silenced in the aberrantly methylated samples. Patients with chromosome rearrangements showed distinct methylation signatures. To establish the role of fusion proteins in the epigenetic profiles, 20 additional samples of human hematopoietic stem/progenitor cells (HSPC) transduced with common fusion genes were studied and compared with patient samples carrying the same rearrangements. The presence of MLL rearrangements in HSPC induced the methylation profile observed in the MLL-positive primary samples. In contrast, fusion genes such as AML1/ETO or CBFB/MYH11 failed to reproduce the epigenetic signature observed in the patients.
Conclusions/significance: Our study provides a comprehensive epigenetic profiling of AML, identifies new clinical markers for cases with a normal karyotype, and reveals relevant biological information related to the role of fusion proteins on the methylation signature.
Conflict of interest statement
Figures
References
-
- Gilliland DG. Hematologic malignancies. Curr Opin Hematol. 2001;8:189–191. - PubMed
-
- Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–6295. - PubMed
-
- Esteller M, Fraga MF, Paz MF, Campo E, Colomer D, et al. Cancer epigenetics and methylation. Science. 2002;297:1807–1808. discussion 1807–1808. - PubMed
-
- Galm O, Herman JG, Baylin SB. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev. 2006;20:1–13. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
