

The PPCD1 mouse: characterization of a mouse model for posterior polymorphous corneal dystrophy and identification of a candidate gene
- PMID: 20808945
- PMCID: PMC2922377
- DOI: 10.1371/journal.pone.0012213
The PPCD1 mouse: characterization of a mouse model for posterior polymorphous corneal dystrophy and identification of a candidate gene
Abstract
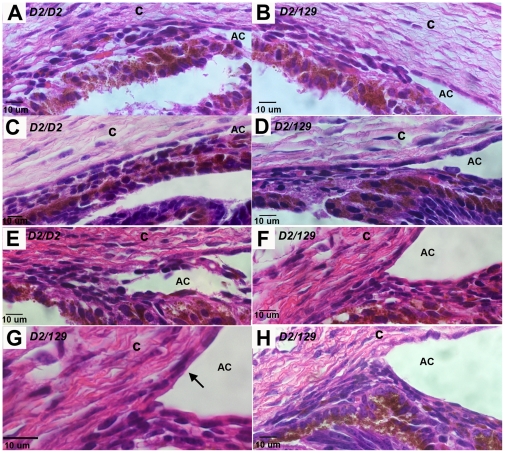
The PPCD1 mouse, a spontaneous mutant that arose in our mouse colony, is characterized by an enlarged anterior chamber resulting from metaplasia of the corneal endothelium and blockage of the iridocorneal angle by epithelialized corneal endothelial cells. The presence of stratified multilayered corneal endothelial cells with abnormal patterns of cytokeratin expression are remarkably similar to those observed in human posterior polymorphous corneal dystrophy (PPCD) and the sporadic condition, iridocorneal endothelial syndrome. Affected eyes exhibit epithelialized corneal endothelial cells, with inappropriate cytokeratin expression and proliferation over the iridocorneal angle and posterior cornea. We have termed this the "mouse PPCD1" phenotype and mapped the mouse locus for this phenotype, designated "Ppcd1", to a 6.1 Mbp interval on Chromosome 2, which is syntenic to the human Chromosome 20 PPCD1 interval. Inheritance of the mouse PPCD1 phenotype is autosomal dominant, with complete penetrance on the sensitive DBA/2J background and decreased penetrance on the C57BL/6J background. Comparative genome hybridization has identified a hemizygous 78 Kbp duplication in the mapped interval. The endpoints of the duplication are located in positions that disrupt the genes Csrp2bp and 6330439K17Rik and lead to duplication of the pseudogene LOC100043552. Quantitative reverse transcriptase-PCR indicates that expression levels of Csrp2bp and 6330439K17Rik are decreased in eyes of PPCD1 mice. Based on the observations of decreased gene expression levels, association with ZEB1-related pathways, and the report of corneal opacities in Csrp2bp(tm1a(KOMP)Wtsi) heterozygotes and embryonic lethality in nulls, we postulate that duplication of the 78 Kbp segment leading to haploinsufficiency of Csrp2bp is responsible for the mouse PPCD1 phenotype. Similarly, CSRP2BP haploinsufficiency may lead to human PPCD.
Conflict of interest statement
Figures







References
-
- Ciralsky J, Colby K. Congenital corneal opacities: a review with a focus on genetics. Semin Ophthalmol. 2007;22:241–246. - PubMed
-
- Cockerham GC, Laver NV, Hidayat AA, McCoy DL. An immunohistochemical analysis and comparison of posterior polymorphous dystrophy with congenital hereditary endothelial dystrophy. Cornea. 2002;21:787–791. - PubMed
-
- Hidayat AA, Cockerham GC. Epithelial metaplasia of the corneal endothelium in Fuchs endothelial dystrophy. Cornea. 2006;25:956–959. - PubMed
-
- Hirst LW, Bancroft J, Yamauchi K, Green WR. Immunohistochemical pathology of the corneal endothelium in iridocorneal endothelial syndrome. Invest Ophthalmol Vis Sci. 1995;36:820–827. - PubMed
-
- Lefebvre V, Sowka JW, Frauens BJ. The clinical spectrum between posterior polymorphous dystrophy and iridocorneal endothelial syndromes. Optometry. 2009;80:431–436. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
