

Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β
- PMID: 20811150
- PMCID: PMC2947222
- DOI: 10.1172/JCI42028
Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β
Abstract
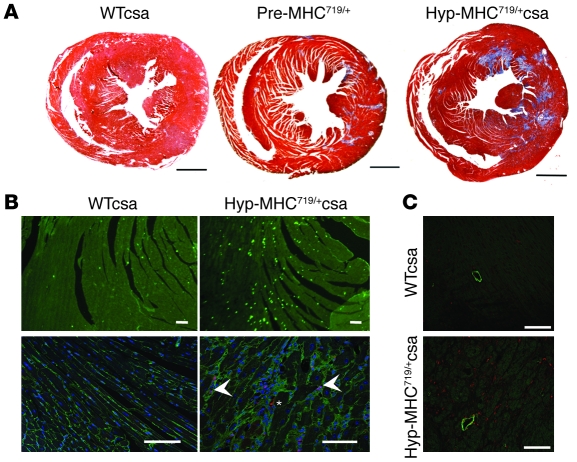
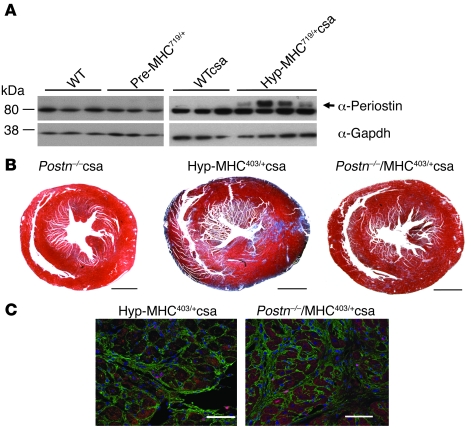
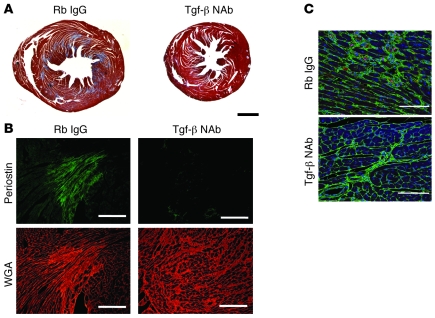
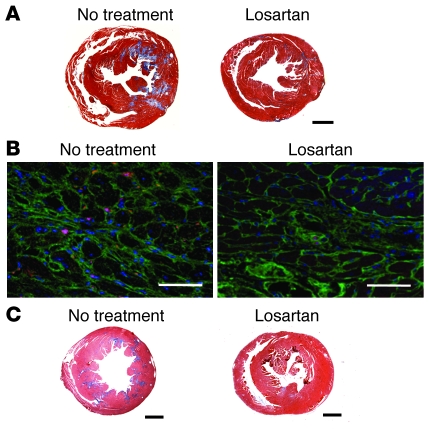
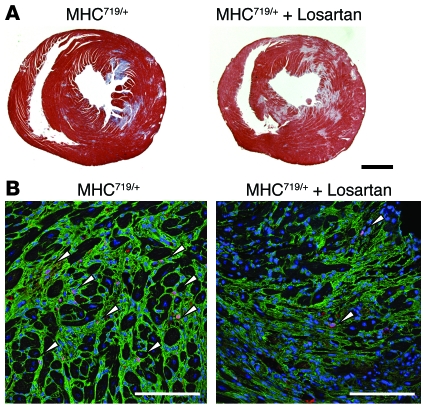
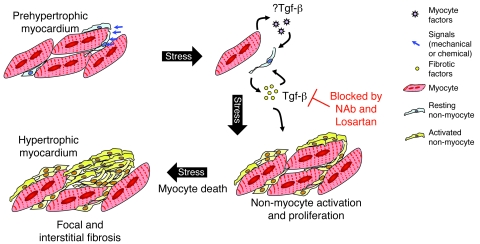
Mutations in sarcomere protein genes can cause hypertrophic cardiomyopathy (HCM), a disorder characterized by myocyte enlargement, fibrosis, and impaired ventricular relaxation. Here, we demonstrate that sarcomere protein gene mutations activate proliferative and profibrotic signals in non-myocyte cells to produce pathologic remodeling in HCM. Gene expression analyses of non-myocyte cells isolated from HCM mouse hearts showed increased levels of RNAs encoding cell-cycle proteins, Tgf-β, periostin, and other profibrotic proteins. Markedly increased BrdU labeling, Ki67 antigen expression, and periostin immunohistochemistry in the fibrotic regions of HCM hearts confirmed the transcriptional profiling data. Genetic ablation of periostin in HCM mice reduced but did not extinguish non-myocyte proliferation and fibrosis. In contrast, administration of Tgf-β-neutralizing antibodies abrogated non-myocyte proliferation and fibrosis. Chronic administration of the angiotensin II type 1 receptor antagonist losartan to mutation-positive, hypertrophy-negative (prehypertrophic) mice prevented the emergence of hypertrophy, non-myocyte proliferation, and fibrosis. Losartan treatment did not reverse pathologic remodeling of established HCM but did reduce non-myocyte proliferation. These data define non-myocyte activation of Tgf-β signaling as a pivotal mechanism for increased fibrosis in HCM and a potentially important factor contributing to diastolic dysfunction and heart failure. Preemptive pharmacologic inhibition of Tgf-β signals warrants study in human patients with sarcomere gene mutations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
