

Emerging principles in protease-based drug discovery
- PMID: 20811381
- PMCID: PMC2974563
- DOI: 10.1038/nrd3053
Emerging principles in protease-based drug discovery
Abstract
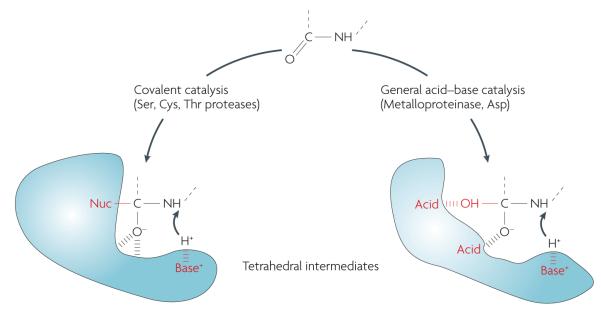
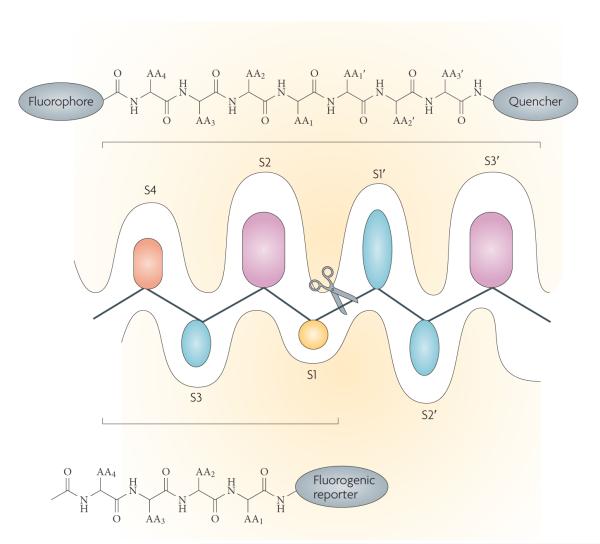
Proteases have an important role in many signalling pathways, and represent potential drug targets for diseases ranging from cardiovascular disorders to cancer, as well as for combating many parasites and viruses. Although inhibitors of well-established protease targets such as angiotensin-converting enzyme and HIV protease have shown substantial therapeutic success, developing drugs for new protease targets has proved challenging in recent years. This in part could be due to issues such as the difficulty of achieving selectivity when targeting protease active sites. This Perspective discusses the general principles in protease-based drug discovery, highlighting the lessons learned and the emerging strategies, such as targeting allosteric sites, which could help harness the therapeutic potential of new protease targets.
Figures
References
-
- Turk B. Targeting proteases: successes, failures and future prospects. Nature Rev. Drug Discov. 2006;5:785–799. - PubMed
-
- Smith CG, Vane JR. The discovery of captopril. FASEB J. 2003;17:788–789. - PubMed
-
- Flexner C, Bate G, Kirkpatrick P. Tipranavir. Nature Rev. Drug Discov. 2005;4:955–956. - PubMed
-
- Melnikova I. The anticoagulants market. Nature Rev. Drug Discov. 2009;8:353–354. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
